
Abstract
Purpose
Modulation of estrogen receptor (ER) plays a central role in selective estrogen receptor modulators (SERMs) molecular mechanism of action, although studies have indicated that additional, non-ER-mediated mechanisms exist. It has been suggested that the induction of oxidative stress by SERM could be one of the non-ER-mediated mechanisms held responsible for their pro-apoptotic role in ER-negative cells. Tumor cells are known for their high requirement of glutamine (Gln) that serves multiple functions within the cells, including nutritional and energy source, as well as one of the precursors for the synthesis of natural antioxidant glutathione (GSH). We hypothesized that one of the mechanisms responsible for ER-independent anti-neoplastic properties of SERMs and also for their adverse side effects could be dependent on the inhibition of Gln uptake.
Methods
Human ER-negative MDA-MB231 breast cancer cells were treated with different doses of Tam and Ral. Gln uptake was monitored by using [3H]Gln assay. The effect of Tam and Ral on Gln transporter ASCT2 expression, glutathione (GSH) levels and cellular proliferation was determined.
Results
Tam and Ral inhibited Gln uptake in a dose-dependent manner through inhibition of ASCT2 Gln transporter. This effect of the anti-estrogens was associated with inhibition of GSH production and apoptosis. Treatment of cells with N-acetyl L-cysteine and 17 beta-estradiol 2 reversed the effects of Ral and Tam.
Conclusions
Our results indicate that one of the mechanisms of action (and possibly some of the side effects) of TAM and RAL is associated with inhibition of cellular Gln uptake, oxidative stress and induction of apoptosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The efficacy of the selective estrogen receptor modulators (SERMs) tamoxifen (TAM) and raloxifene (RAL) has been attributed to the induction of tumor cell growth arrest and apoptosis by inhibition of estrogen receptor (ER) signaling [1, 2]. Although SERMs have widespread clinical use, it is clear that not all of their effects can be attributed to the competitive interaction with the estrogen receptor. Experimental studies with TAM found growth inhibition and apoptosis in ER(-) cell lines [3, 4]. Responses to Tam have been observed in cancers not derived from estrogen-sensitive tissues, such as glioma [5, 6], melanoma[7], pancreatic cancer [8] and other malignancies [9]. Tam therapy has also been used for treatment of a number of other diseases such as osteoporosis [10, 11], atherosclerosis [12, 13], rheumatoid arthritis and other autoimmune diseases [14]. Tam has been shown to be effective in the treatment of mania in patients with bipolar disorder [15]. The cytotoxic effect of SERMs is believed to be due to a combination of genomic (ER-mediated) and non-genomic (non-ER-mediated or other signaling pathways) mechanisms. These include modulation of signaling proteins such as protein kinase C (PKC), calmodulin, transforming growth factor-β (TGF-β), caspases, mitogen-activated protein kinases (MAPK) and the protooncogene c-myc [reviewed in 16]. Oxidative stress, mitochondrial permeability transition (MPT) and ceramide generation also play important roles in TAM-induced apoptosis [15].
Glutamine (Gln) is an essential nutrient for cell growth and viability [17]. In general the functions of Gln within the cell include its roles in nitrogen transport, maintaining cellular redox state, as a metabolic intermediate and as an energy source [18]. Gln via glutamate, together with glycine and cysteine is a precursor for the synthesis of glutathione (GSH), the major endogenous antioxidant in mammalian cells, which protects them from oxidative injury [19]. Gln is utilized directly for protein synthesis and serves as a precursor in the synthesis of other amino acids [20]. Some tumor cells have an absolute requirement for Gln as a growth substrate, a precursor for both DNA- and protein synthesis, as well as a respiratory substrate [21–23]. DeBerardinis et al. [24] reported that glioma cells can exhibit Gln uptake and metabolism that exceeds the cell’s use of Gln for protein and nucleotide biosynthesis. This high rate of glutaminolysis was found to be beneficial because it provided the cell a high rate of NADPH production that was used to fuel lipid and nucleotide biosynthesis. In cancer patients, some tumors have been reported to consume such an abundance of Gln that they depress plasma Gln levels [25, 26].
Gln transport across cell membrane is mediated by several transport systems [27], the predominant one for human cancer cells being ASCT2 [28]. Studies have found that Gln availability regulated the expression of ASCT2 transporter in hepatoma cells [29]. The aim of the present work was to identify an ER-independent pathway that could explain, at least in part, the antineoplastic activity of TAM reported in ER-negative cells and ER-poor tumors. We hypothesized that one of the mechanisms responsible for ER-independent anti-neoplastic properties of SERMs and also for their adverse side effects could be dependent on the inhibition of Gln uptake.
Materials and methods
Cell cultures, chemicals and treatment
Human ER-negative breast cancer cell line MDA-MB-231 at passage 26 was obtained from the ATCC (Manassas, VA) and was maintained in Dulbecco’s modified essential medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin/streptomycin (P/S), and 2 mM glutamine. Tamoxifen citrate, raloxifene hydrochloride, 17β-estradiol 2 (E2), sodium selenite (Se), retinyl hydrochloride (Vit A), α-tocoferol (Vit E) and N-acetyl-cysteine (NAC) were purchased from Sigma–Aldrich (St Louis, MO). Rabbit anti-rat ASCT2 antibody was from Chemicon. International (Temecula, CA) and anti-rabbit IgG were from Santa Cruz Biotech (Santa Cruz, CA). Radiolabeled [3H]Gln was purchased from Amersham Pharmacia Biotech (Piscataway, NJ).
Cell proliferation/viability assay
Cells (1 × 104 cells/well) were plated in quadruplicate in a 96-well plate in 100 μl complete medium and after attachment for 24 h were cultured overnight in serum-free medium, followed by treatment with increasing concentrations of Tam or Ral (5–25 μM) for 48 h at 37°C under 5% carbon dioxide (CO2)/95% air in a high humidity atmosphere. The effect of NAC, E2, Vit E, Vit A and selenium was determined as follows: cells were plated in 96 well plate in triplicate (1 × 104 cells/well) and incubated 24 h at 37°C and 5% CO2 in complete medium. The medium was replaced with serum-free DMEM with each of NAC (5 mM), E2 (10 nM), Vit E (100 μM), Vit A (1 μM) and selenium (30 nM) and incubated for 1 h before challenged with Ral (25 μM) or Tam (μM) for 48 h. Proliferation was assayed using a modified MTS-tetrazolium (MTS) assay with CellTiter 96 Aqueous reagent (Promega, Madison, WI) and measurement of absorbance at 490 nm, according to the manufacturer’s protocol. Inhibition of proliferation was calculated as percentage of the control cultures that were not treated with Tam or Ral. All experiments were repeated at least three times. A paired t test with P < 0.05 was used to establish statistically significant differences between treatment and control.
Glutathione measurement
Cells (1 × 104 cells/well) were attached in quadruplicate in a 96-well plate in 100 μl complete medium and cultured overnight in serum-free medium before the treatment with increasing concentrations of Tam or Ral (5–25 μM) for 48 h at 37°C under 5% carbon dioxide (CO2). The effect of NAC, E2, Vit E, Vit A and selenium was determined by preincubation the cells for 1 h with NAC (5 mM), E2 (10 nM), Vit E (100 μM), Vit A (1 μM) and selenium (30 nM), followed by Ral (25 μM) or Tam (μM) for 48 h. The amount of total GSH released in the medium was estimated by measuring the absorbance at 405 nm (GSH assay kit; Cayman Chemical, Ann Arbor, MI) using a microplate reader (Bio-Rad Laboratories, Hercules, CA). Pure oxidized GSH (GSSG) was used to obtain a standard curve. The results were expressed as nmol/mg protein.
Glutamine uptake assay
Cells were plated in 12-well plates (1 × 105/well) and after attachment for 24 h the medium was replaced with fresh Gln-free medium and incubated overnight. The next day, the medium was replaced with medium containing 2 mM Gln, 5 μCi [3H]Gln and various concentrations of Ral and Tam (5–25 μM) and the cells were incubated at 37°C, 5% CO2 for 45 min. The Gln uptake was terminated by three washes with cold PBS and drying at room temperature for 30 min. The cells were dissolved with 1 ml/well buffer containing 0.2N NaOH and 0.2% SDS, followed by the addition of 100 μl/well 2N HCl. The protein was measured in the lysate, and the radioactivity was measured in 5 ml scintillation cocktail with LKB Wallac 1219 liquid scintillation counter. The results are expressed as cpm/mg protein.
DNA fragmentation assay
After treatment, the cells were harvested by trypsinization and centrifuged at 1,000g for 5 min, washed once with PBS and the cell pellet was resuspended in a lysis buffer containing 10 mM Tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, and 0.5% Triton X-100. Cell lysate was left on ice for 30 min. DNA was extracted by adding an equal volume of neutral phenol:chloroform:isoamyl alcohol mixture (pH 8.0) and precipitated with 0.1 volume of 5 M sodium chloride and 2 volumes of 100% ethanol at −20°C overnight. The DNA sample was dissolved in TE buffer (10 mM Tris, pH 8.0 and 1 mM EDTA) and treated with 1 mg/ml RNase at 37°C for 2 h. DNA fragments were resolved by electrophoresis in a 1.5% agarose gel and visualized by ethidium bromide staining.
Western blotting
Cells (5 × 106 in 100 mm plates) were treated as indicated, harvested and sonicated in cold cell lysis buffer (Cell Signaling Tech, Danvers, MA). Protein concentration was measured using Bio-Rad protein assay (Richmond, VA). Fifty microgram of the total cytosolic protein were resolved in 10% SDS–PAGE and transferred to nitrocellulose membrane (Millipore). The membrane was blocked with 5% non-fat dry milk in TBS for 1 h at room temperature and then incubated with anti-ASCT2 antibody (1:800 in TBS-T containing 5% milk) overnight at 4°C. After washing the blots were incubated for 2 h at 4°C in ant-rabbit IgG (1:2000). The protein was visualized using enhanced chemiluminescence system (Amersham). The equal loadings were controlled by staining with Ponso S and reprobing the membranes with tubulin.
Statistical analysis
Comparisons between the groups were performed by a one-way analysis of ANOVA using statistical software StatView for Windows. All data was expressed as mean ± SE. Results with P < 0.05 were considered statistically significant.
Results
Cell proliferation/apoptosis and GSH synthesis
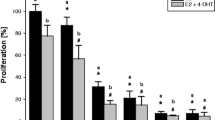
Treatment with Tam and Ral induced a dose-dependent reduction in cell survival (Fig. 1a) and down-regulation of GSH synthesis (Fig. 1b) of human ER-negative MDA-MB231 breast cancer cells. The most prominent effect of both Tam and Ral was found at a concentration of 25 μM with a decrease in the number of live cells by more than 60%. This concentration of both Tam- and Ral (25 μM)-induced DNA fragmentation (Fig. 1c) suggestive for the occurrence of apoptosis.

Dose-dependent inhibition of cell proliferation (a) and GSH synthesis (b) and induction of apoptosis (c) in MDA-MB 231 cells by Ral and Tam. Cells were treated with various concentrations (5–25 μM) of Tam or Ral for 48 h as described in “Materials and methods”. Cell survival was determined using MTS assay (a) and GSH released in the medium was determined using GSH assay (b). After treatment with Tam and Ral (25 μM) for 48 h, the cells were harvested, DNA was extracted and analyzed by agarose gel electrophoresis (c). C1, untreated control; C2, cells treated with 25 μM Tam; C3, cells treated with 25 μM Ral. Three independent experiments showed similar results. Data are mean ± SE. P < 0.05
Tam- and Ral-induced GSH depletion and inhibition of cell proliferation are reversed by NAC and E2
Treatment with 25 μM Tam or Ral for 48 h resulted in a significant GSH depletion (Fig. 2a) that was reversed by a pretreatment with NAC or E2, but not with Vit E, Vit A or selenium. The same effect of these antioxidants was found also on cell proliferation (Fig. 2b).

The down-regulation of GSH (a) and suppression of cellular proliferation (b) of MDA-MB 231 cells by 25 μM Ral or Tam was reversed by pretreatment with NAC and E2. Cells were pretreated for 1 h with the indicated antioxidants and challenged with Ral or Tam for 48 h. Cell survival was determined by using MTS assay and GSH by a specific assay. P < 0.05
Gln uptake
In order to assess the effect of Ral and Tam (concentrations range 5–25 μM) on the uptake of Gln, MDA-MB 231 cells were incubated for 45 min in a medium containing 5 μCi [3H]Gln. The results showed that 5 μM of both Ral and Tam caused a sharp decrease in Gln uptake that continued to decrease with the increase in Ral and Tam concentration (Fig. 3.)

Tam and Ral inhibit Gln-uptake. Cells were incubated for 45 min in a medium containing 2 mM Gln, 5 μCi [3H]Gln and various concentrations of Ral and Tam (5–25 μM). Gln uptake was assessed by measuring the radioactivity with liquid scintillation counter. P < 0.05
Protein expression of Gln transporter ASCT2
In order to clarify the biochemical mechanism underlying Tam- and Ral-induced inhibition of Gln uptake, we examined the protein expression of Gln transporter ASCT2. MDA-MB 231 cells were treated with Ral and Tam (25 μM), and 50 μg of the total cytosolic protein of untreated cells and cells treated with Ral and Tam were examined for the protein expression of ASCT2. The results showed that both Ral and Tam down-regulated the protein expression of ASCT2 (Fig. 4).

Ral and Tam inhibited protein expression of Gln transporter ASCT2. The presence of ASCT2 protein was determined by western blot analysis. The equal loadings were controlled by staining with Ponso S and reprobing the membranes with tubulin. a Untreated cells; b cells treated with 25 μM Ral; c cells treated with 25 μM Tam
Discussion
Several large clinical trials have demonstrated the efficacy of the SERM, specifically of Tam in the adjuvant therapy of breast cancer. Although the effect of TAM in terms of recurrence-free survival was detected in ER-dependent tumors, a significant favorable effect was detected also in ER-poor cancers [30]. The present study has shown that both anti-estrogens Tam and Ral down-regulated GSH production and induced apoptosis in ER-negative breast cancer cells grown under stressed (serum free) conditions. These effects of Tam and Ral were associated with inhibition of Gln uptake in a dose-dependent manner through an inhibition of ASCT2 Gln transporter. Treatment of cells with NAC and E2 reversed the effects of Ral and Tam.
Several in vitro properties of SERMs were suggested to be responsible for their pro-apoptotic properties in ER-negative cells including calcium channel–blocking activity, inhibition of protein kinase C [31], up-regulation of c-myc expression [32], ceramide generation [33], activation of caspases and JNK and p38 mitogen-activated protein kinases (MAPK) [34]. It has been suggested that Tam exerts a direct regulatory effect on the transcription of TGF-β. Studies showed that prolonged incubation of breast cancer cells with Tam resulted in accumulation of TGF-β mRNA and protein, as well as increased biological activity of TGF-β secreted in the cell culture media [35, 36]. The addition of an anti-TGF-β antibody inhibited the induction of apoptosis by TAM. Brunner et al. [37] reported that treatment with Tam significantly reduced the concentration of insulin-like growth factor II (IGF-II) in T61 human breast cancer xenografts. Reactive oxygen species (ROS) have been demonstrated to play an essential role in apoptosis induced by TAM in ER-negative human cancer cells [38, 39]. For example, Ferlini et al. [4] showed that Tam-induced apoptosis in ER-negative models resulted from ROS generation and thiol depletion in a dose-dependent manner. Pretreatment of ER-negative breast cancer cells with the antioxidant Vit E abrogated JNK activation in TAM-treated cells, suggesting the significant role of the oxidative stress in TAM-induced cellular apoptosis [24]. In fact, several studies showed that the oxidative stress induced by SERM is one of the ER-independent mechanisms of action of these drugs [40–42]. The major anti-oxidant defense system of the cell, GSH is a tripeptide (γ-glutamyl-cysteinyl-glycin) synthesized in a two-step reaction of combination of glutamate and cysteine, followed by the addition of glycine [43, 44]. It is generally accepted that the availability of cysteine [45] and Gln [46] (as a precursor of glutamate) controls the rate of this reaction. Gln supports the intracellular pool of glutamate, avoiding its depletion and the depletion of GSH. In vitro and in vivo studies have shown that NAC is chemically similar to cysteine and acts as a cysteine prodrug and a GSH precursor [47].
The results from this study showed that Ral and Tam inhibition of GSH synthesis of MDA-MB 231 cells was reversed by the addition of NAC (but not by other antioxidants), suggesting that GSH down-regulation might depend on Gln availability. The examination of the effect of Ral and Tam on Gln uptake showed a significant inhibition of Gln uptake. Studies have found that cancer cells displayed enhanced and altered channeling of amino acids into select metabolic pathways, often in concert with the aerobic glycolysis characteristic of tumors [48, 49]. Solid tumors are often poorly vascularized, so they must have efficient mechanisms for extracting plasma amino acids in order to compete with the host tissues [50]. As a result, cancer cell amino acid transporters with properties that impart growth and survival advantages are selected for and expressed at higher levels compared to the parent tissue [19]. Amino acid transport across the plasma membrane in mammalian cells is mediated by different transport systems, such as Na+-dependent systems A, ASC, Na+-independent system L and N system [51]. ASCT2 transporter has a high affinity for small neutral amino acids, such as glutamine which is avidly consumed by tumors. Current evidence suggests that ASCT2 is highly expressed in a variety of cancerous tissues, such as breast [52], colon, liver and other cancers; and therefore, it has been suggested to play an important role in the carcinogenesis [19, 37]. The importance of ASCT2 transporter in hepatoma cell proliferation has been demonstrated by Fuchs et al. [53] who showed that attenuation of ASCT2 expression by inducible antisense RNA in a number of human hepatoma cell lines led to cell death via apoptosis. Bungard and McGivan [20] found that both the expression of the glutamine transporter ASCT2 and the activity of the ASCT2 promoter in the human hepatoma cell line HepG2 were dependent upon Gln availability. The results from the present study showed that Ral- and Tam-induced inhibition of Gln uptake by MDA-MB-231 breast cancer cells were associated and probably resulted from inhibition of ASCT2 protein expression.
In our experimental model, the inhibitory effects of Ral and Tam on GSH synthesis and proliferation of ER-negative cells were reversed also by 17β-estradiol, suggesting the presence of ER-independent mechanism of action of estrogens. Recent study by Wang et al. [54] showed that 17β-, 17α- and ent-E2 protected human neuroblastoma SK-N-SH cells against H2O2-induced oxidative stress by inhibition of lipid peroxidation, alleviated intracellular calcium elevation, attenuated ATP depletion and subsequently enhanced cell survival. Furthermore, the ER antagonist ICI 182,780 did not block the effects of 17-E2 but increased cell survival and blunted intracellular calcium increase induced by H2O2.
The results from this study indicate that one of the mechanisms of action (and possibly some of the side effects) of anti-estrogens TAM and RAL is associated with inhibition of cellular GLN uptake and subsequent oxidative stress and induction of apoptosis. These data contribute to the knowledge of mechanism of action of two of the most widely used in the clinical practice SERM.
References
Love RR (1989) Tamoxifen therapy in primary breast cancer: biology, efficacy, and side effects. J Clin Oncol 7:803–815
Jaiyesimi IA, Buzdar AU, Decker DA, Hortobagyi GN (1995) Use of tamoxifen for breast cancer: twenty-eight years later. J Clin Oncol 13:513–529
Perry RR, Kang Y, Greaves B (1995) Effects of tamoxifen on growth and apoptosis of oestrogen-dependent and -independent human breast cancer cells. Ann Surg Oncol 2:238–245
Ferlini C, Scambia G, Marone M, Distefano M, Gaggini C, Ferrandina G, Fattorossi A, Isola G, Benedetti Panici P, Mancuso S (1999) Tamoxifen induces oxidative stress and apoptosis in oestrogen receptor-negative human cancer cell lines. Br J Cancer 79:257–263
Corti PS (1993) Apuzzo MLJ: Clinical and radiographic response in a minority of patients with recurrent malignant gliomas treated with high-dose tamoxifen. Neurosurgery 32:485–490
Couldwell WT, Hinton DR, He S, Chen TC, Sebat I, Weiss MH, Law RE (1994) Protein kinase C inhibitors induce apoptosis in human malignant glioma cell lines. FEBS Lett 345:43–46
McClay EF, Albright KD, Jones JA, Christen RD, Howell SB (1993) Tamoxifen modulation of cisplatin sensitivity in human malignant melanoma cells. Cancer Res 53:1571–1576
Taylor OM, Benson EA, McMahon MJ (1993) Clinical trial of tamoxifen in patients with irresectable pancreatic adenocarcinoma. The Yorkshire Gastrointestinal Tumor Group. Br J Surg 80:384–386
Gelmann EP (1997) Tamoxifen for the treatment of malignancies other than breast and endometrial carcinoma. Semin Oncol 24:S1-65–S1-70
Ward RL, Morgan G, Falley D, Kelly PJ (1993) Tamoxifen reduces bone turnover and prevents lumbar spine proximal femoral bone loss in early postmenopausal women. Bone Miner 22:87–94
Recker R (1993) Clinical review 41: current therapy for osteoporosis. J Clin Endocrinol Metab 76:14–16
Grainger DJ, Witchell CM (1995) Metcalfe JC: Tamoxifen elevates transforming growth factor-ß suppresses diet-induced formation of lipid lesions in mouse aorta. Nat Med 1:1067–1073
Reckless J, Metcalfe JC, Grainger DJ (1997) Tamoxifen decreases cholesterol seven-fold and abolishes lipid lesion development in apolipoprotein E knock out mice. Circulation 95:1542–1548
Sthoeger Z, Dayan M, Zinger H, Kalush F, Mor G, Zlatman YA, Kohen F, Mozes E (1997) Treatments with tamoxifen and an antiestradiol antibody have beneficial effects on experimental SLE via cytokine modulation. Ann N Y Acad Sci 815:367–368
Yildiz A, Guleryuz S, Ankerst DP, Ongür D, Renshaw PF (2008) Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry 65:255–263
Mandlekar S, Kong A-NT (2001) Mechanisms of tamoxifen-induced apoptosis. Apoptosis 6:469–477
Eagle H (1955) Nutrition needs of mammalian cells in tissue culture. Science 122:501–514
Labow BI, Souba WW (2000) Glutamine. W J Surgery 24:1503–1513
Meister A, Anderson MA (1983) Glutathione. Annu Rev Biochem 52:711–721
Abcouwer SF, Bode BP, Souba WW (1996) Glutamine as a metabolic intermediate. In: Fisher JE (ed) Nutrition and metabolism in the surgical patient. Little Brown, Boston, pp 353–384
Coles NW, Johnstone RM (1962) Glutamine metabolism in Ehrlich ascites-carcinoma cells. Biochem J 83:284–291
Souba WW (1993) Glutamine and cancer. Ann Surgery 218:715–728
Mates JM, Perez-Gomez C, Nunez DC, Asenjo IM, Marquez J (2002) Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int J Biochem Cell Biol 34:439–458
DeBerardinis RJ, DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104:19345–19350
Klimberg VS (1996) Glutamine, cancer, and its therapy. Am J Surgery 172:418–424
Chen MK, Espat NJ, Bland KI, Copeland EM III, Souba WW (1993) Influence of progressive tumor growth on glutamine metabolism in skeletal muscle and kidney. Ann Surg 217:655–666
Bode BP (2001) Recent molecular advances in mammalian glutamine transport. J Nutr 131(Suppl):2475S–2487S
Fuchs BC, Bode BP (2005) Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Sem Cancer Biol 15:254–266
Bungard CI, McGivan JD (2004) Glutamine availability up-regulates expression of the amino acid transporter protein ASCT2 in HepG2 cells and stimulates the ASCT2 promoter. Biochem J 382:27–32
Jaiyesimi IA, Buzdar AU, Decker DA, Hortobagyi GN (1995) Use of tamoxifen for breast cancer: twenty-eight years later. J Clin Oncol 13:513–529
O’Brian CA, Housey GM, Weinstein IB (1988) Specific and direct binding of protein kinase C to an immobilized tamoxifen analogue. Cancer Res 48:3626–3629
Kang Y, Cortina R, Perry RR (1996) Role of c-myc in tamoxifen induced apoptosis estrogen-independent breast cancer cells. J Natl Cancer Inst 88:279–284
Maurer BJ, Metelitsa LS, Seeger RC, Cabot MC, Reynolds CP (1999) Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)- retinamide in neuroblastoma cell lines. J Natl Cancer Inst 91:1138–1146
Mandlekar S, Yu R, Tan TH, Kong AN (2000) Activation of caspase-3 and c-Jun NH2-terminal kinase-1 signaling pathways in tamoxifen-induced apoptosis of human breast cancer cells. Cancer Res 60:5995–6000
Koli KM, Ramsey TT, Ko Y, Dugger TC, Brattain MG, Arteaga CL (1997) Blockade of transforming growth factor-beta signaling does not abrogate antiestrogen-induced growth inhibition of human breast carcinoma cells. J Biol Chem 272:8296–8302
Perry RR, Kang Y, Greaves BR (1995) Relationship between tamoxifen-induced transforming growth factor beta 1 expression, cytostasis and apoptosis in human breast cancer cells. Br J Cancer 72:1441–1446
Brünner N, Yee D, Kern FG, Spang-Thomsen M, Lippman ME, Cullen KJ (1993) Effect of endocrine therapy on growth of T61 human breast cancer xenografts is directly correlated to a specific down-regulation of insulin-like growth factor II (IGF-II). Eur J Cancer 29A(4):562–569
Ferlini C, Scambia G, Marone M, Distefano M, Gaggini C, Ferrandina G, Fattorossi A, Isola G, Benedetti Panici P, Mancuso S (1999) Tamoxifen induces oxidative stress and apoptosis in oestrogen receptor-negative human cancer cell lines. Br J Cancer 79:257–263
Hayon T, Dvilansky A, Oriev L, Nathan I (1999) Non-steroidal antiestrogens induce apoptosis in HL60 and MOLT3 leukemic cells; involvement of reactive oxygen radicals and protein kinase C. Anticancer Res 19:2089–2093
Mandlekar S, Kong AN (2001) Mechanisms of tamoxifen-induced apoptosis. Apoptosis 6:469–477
Dietze EC, Caldwell LE, Grupin SL, Mancini M, Seewaldt VL (2001) Tamoxifen but not 4-hydroxytamoxifen initiates apoptosis in p53(–) normal human mammary epithelial cells by inducing mitochondrial depolarization. J Biol Chem 276:5384–5394
Nazarewicz RR, Zenebe WJ, Parihar A, Larson SK, Alidema E, Choi J, Ghafourifar P (2007) Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res 67:1282–1290
Kosower NS, Kosower EM (1978) The glutathione status of cells. Intl Rev Cytology 54:109–156
Kehrer JP, Lund LG (1994) Cellular reducing equivalents and oxidative stress. Free Rad Biol Med 17:65–75
Griffith OW (1999) Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med 27(9–10):922–935
Amores-Sanchez MI, Medina MA (1999) Glutamine, as a precursor of glutathione, and oxidative stress. Mol Genet Metabol 67:100–105
Zafarullah M, Li WQ, Sylvester J, Ahmad M (2003) Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 60:6–20
Baggetto LG (1992) Deviant energetic metabolism of glycolytic cancer cells. Biochimie 74:959–974
Mazurek S, Eigenbrodt E (2003) The tumor metabolome. Anticancer Res 23:1149–1154
Medina MA, Sanchez-Jimenes F, Marquez J, Rodruguez Quesada A, Nunez de Castro I (1992) Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem 113:1–15
Palacin M, Estevez R, Bertran J, Zorzano A (1998) Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 78:969–1054
Collins CL, Wasa M, Souba WW, Abcouwer SF (1998) Determinants of glutamine dependence and utilization by normal and tumor-derived breast cell lines. J Cell Physiol 176:166–178
Fuchs BC, Perez JC, Suetterlin JE, Chaudhry SB, Bode BP (2004) Inducible antisense RNA targeting amino acid transporter ATB0/ASCT2 elicits apoptosis in human hepatoma cells. Am J Physiol Gastrointest Liver Physiol 286:G467–G478
Wang X, Dykens JA, Perez E, Liu R, Yang S, Covey DF, Simpkins JW (2006) Neuroprotective effects of 17beta-estradiol and nonfeminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH cells. Mol Pharmacol 70:395–404
Acknowledgments
This work was supported by VA Merit Review Award to VS Klimberg.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Todorova, V.K., Kaufmann, Y., Luo, S. et al. Tamoxifen and raloxifene suppress the proliferation of estrogen receptor-negative cells through inhibition of glutamine uptake. Cancer Chemother Pharmacol 67, 285–291 (2011). https://doi.org/10.1007/s00280-010-1316-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-010-1316-y