
Abstract
Purpose
The development of multi-drug resistance (MDR) due to the expression of members of the ATP binding cassette (ABC) transporter family is a major obstacle in cancer treatment. The broad range of substrate specificities associated with these transporters leads to the efflux of many anti-cancer drugs from tumour cells. Therefore, the development of new chemotherapeutic agents that are not substrates of these transporters is important. We have recently demonstrated that some members of a novel series of pyrrolo-1,5-benzoxazepine (PBOX) compounds are microtubule-depolymerising agents that potently induce apoptosis in several cancer cell lines and impair growth of mouse breast tumours. The aim of this current study was to establish whether PBOXs were capable of inducing apoptosis in cancer cells expressing either P-glycoprotein or breast cancer resistance protein (BCRP), two of the main ABC transporters associated with MDR.
Methods
We performed in vitro studies to assess the effects of PBOXs on cell proliferation, cell cycle and apoptosis in human cancer cell lines and their drug-resistant substrains expressing either P-glycoprotein or BCRP. In addition, we performed a preliminary molecular docking study to examine interactions between PBOXs and P-glycoprotein.
Results
We established that three representative PBOXs, PBOX-6, -15 and -16 were capable of inducing apoptosis in drug-resistant HL60-MDR1 cells (expressing P-glycoprotein) and HL60-ABCG2 cells (expressing BCRP) with similar potencies as in parental human promyelocytic leukaemia HL60 cells. Likewise, resistance to PBOX-6 and -16 was not evident in P-glycoprotein-expressing A2780-ADR cells in comparison with parent human ovarian carcinoma A2780 cells. Finally, we deduced by molecular docking that PBOX-6 is not likely to form favourable interactions with the substrate binding site of P-glycoprotein.
Conclusion
Our results suggest that pro-apoptotic PBOX compounds may be potential candidates for the treatment of P-glycoprotein- or BCRP-associated MDR cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Some pyrrolo-1,5-benzoxazepine (PBOX) compounds have been identified as novel microtubule-depolymerising agents [1] that possess the ability to potently induce apoptosis in several cancerous cell lines including promyelocytic leukaemia HL60 cells, Jurkat T-lymphoma cells, Hut-78 lymphoma cells, T cell leukaemia CEM cells, highly resistant chronic myeloid leukaemia (CML) K562 cells and breast carcinoma MCF-7 cells [2–6] with minimal toxicity to normal peripheral blood mononuclear cells (PBMCs) [6] or bone marrow cells [7]. We have also reported that PBOXs displayed anti-angiogenic properties in vitro [8]. Furthermore, we have demonstrated impaired growth of tumours in a mouse 4T1 breast carcinoma tumour model [9] and apoptosis in ex vivo patient samples [7]. Therefore, PBOX compounds may prove useful novel anti-cancer agents.
A frequent problem in cancer chemotherapy is the development of multi-drug resistance (MDR) which renders tumours unresponsive to a diverse array of compounds. There are a number of mechanisms by which a cell can acquire MDR, such as a defective apoptotic pathway [10], increasing the activity of detoxifying and DNA repair enzymes [11, 12] and decreasing activity of topoisomerase II [13], however, one of the most predominant causes is due to over-expression of members of the ATP-binding cassette (ABC) transporter family. This is a group of plasma membrane glycoproteins that actively extrude a broad range of substrates from cells. They are predominantly found in areas such as the epithelial cells of the intestine, proximal tubule cells of the kidney, hepatocytes, capillary epithelial cells of the blood–brain barrier, the placenta and in various stem cells where the physiological role of these ATPase efflux “pumps” is to protect cells from damage by rapidly extruding xenobiotics [14–16]. Unfortunately, their unwanted expression in tumour cells can lead to the extrusion of substrate cytotoxic anti-cancer agents preventing their accumulation and hence leading to chemotherapy resistance in these cells.
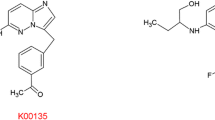
There are three main ABC transporters specifically associated with MDR, these are multi-drug resistance protein-1 (MDR1) also know as P-glycoprotein or ABCB1 [17], multi-drug resistance-associated protein-1 (MRP1) or ABCC1 [18] and the half transporter ABCG2 or breast cancer resistance protein (BCRP) [19, 20]. Some of the key anti-cancer agents known to be substrates of these transporters include microtubule-targeting agents (MTAs) such as paclitaxel and vincristine, anthracyclin antibiotics (e.g. adriamycin), anthracenes (e.g. mitoxantrone), topoisomerase I inhibitors such as topotecan, camptothecin and the active metabolite of irinotecan, SN-38, cyclin-dependent kinase inhibitors (e.g. flavopiridol) and anti-folates (e.g. methotrexate) [14, 21–23]. In addition, a number of other clinically important drugs such as HIV-protease inhibitors, antibiotics, antidepressants, antiepileptics and analgesics are also substrates of the ABC transporters [14]. Since the use of chemical inhibitors of P-glycoprotein or BCRP to block drug efflux have proved unsuccessful clinically, possibly due to the intrinsic toxicity of the actual inhibitors and unwanted inhibition of transporters residing in physiological regions such as the blood–brain barrier leading to altered distribution of the simultaneous administered chemotherapeutic agents [24], the development of novel chemotherapeutic agents that are not substrates of the MDR-ABC transporters has been postulated as the best method to overcome ABC transporter-associated MDR. Therefore, the purpose of this study was to investigate whether representative pro-apoptotic PBOX compounds, PBOX-6, -15 and -16 (Fig. 1), are potential candidates for the treatment of multidrug-resistant cancers caused by the expression of P-glycoprotein or BCRP.

Chemical structure of microtubule-targeting agents PBOX-6, PBOX-15 and PBOX-16
Herein, we demonstrated that PBOXs were capable of inducing cell cycle arrest and apoptosis in MDR leukaemia cells, HL60-MDR1 (expressing P-glycoprotein) and HL60-ABCG2 (expressing BCRP) with similar potencies as in the ABC transporter-negative parental human promyelocytic leukaemia HL60 cell line. Similarly, we determined that P-glycoprotein-expressing A2780-ADR cells did not display resistance to PBOXs compared to the parental human ovarian carcinoma A2780 cell line. Furthermore, through a preliminary molecular docking study, we deduced that PBOXs do not appear to form favourable interactions with the P-glycoprotein substrate binding site. Together, our findings suggested that whilst the efficacy of many common anti-cancer agents may be reduced by the expression of P-glycoprotein or BCRP, the efficacy of PBOX-6, -15 and -16 was not. This indicated that pro-apoptotic PBOX compounds have the potential to treat P-glycoprotein- and BCRP-associated MDR cancers.
Materials and methods
Unless otherwise stated, chemicals were obtained from Sigma–Aldrich (Poole, Dorset, UK), and tissue culture vessels were sourced from Greiner Bio-One GmbH (Frickenhausen, Germany).
Cell culture
Human promyelocytic leukaemia HL60 cells and their drug-resistant substrains HL60-MDR1 and HL60-ABCG2 were provided by Prof. Balazs Sarkadi’s research group. The HL60-MDR1 cells had been drug-selected by chronic exposure to adriamycin, while the HL60-ABCG2 cells had been retrovirally transduced then further selected by exposure to mitoxantrone [25]. Human ovarian carcinoma A2780 cells and their adriamycin-resistant substrain A2780-ADR were obtained from Prof. Robert Brown, University of Glasgow, UK. All cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium enhanced with GlutaMAX-I and supplemented with 10% foetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin (all from Gibco, Invitrogen, Carlsbad, CA, USA). Cells were maintained in a humidified incubator at 37°C in 5% CO2. A2780 cells were subcultured by trypsinisation upon reaching ~80% confluency.
Reagents
The pyrrolo-1,5-benzoxazepine compounds, 7-[(N,N-dimethylcarbamoyl)oxy]-6-(naphth-1-yl)pyrrolo[2,1-d][1,5]benzoxazepine (PBOX-6), 4-acetoxy-5-(1-(naphthyl)naphtho[2,3-b]pyrrolo[2,1-d][1,4]oxazepine (PBOX-15) and 4-[(N,N-dimethylcarbamoyl)oxy]-5-(naphth-1-yl)naphtho[2,3-b]pyrrolo[1,2-d][1,4]oxazepine (PBOX-16) were synthesised as described previously [26] and dissolved in ethanol. Their chemical structure is shown in Fig. 1. 7-Ethyl-10-Hydroxycamptothecin (SN-38) (kindly supplied by Prof. Patrick Johnston, Queen’s University, Belfast, Northern Ireland) was dissolved in DMSO. Paclitaxel and vincristine were dissolved in DMSO or sterile water, respectively. Once dissolved in the relevant solvent, all compounds were stored at −20°C.
Cell proliferation
Cell proliferation was monitored using AlamarBlue™ dye (BioSource, Invitrogen, Carlsbad, CA, USA) which changes to a fluorescent state in the reduced environment of living cells. Cells (25,000 cells/well) were seeded on 96-well plates for 24 h, then treated with a range of concentrations of PBOX-6, PBOX-15, PBOX-16, paclitaxel, vincristine or SN-38 for 72 h. AlamarBlue™ (final concentration 10% (v/v)) was added and incubated at 37°C. Fluorescence was measured at an excitation wavelength of 544 nm and an emission wavelength of 590 nm using a SpectraMax Gemini spectrofluorometric plate reader (Molecular Devices, Sunnyvale, CA). The results were expressed as the percentage cell viability relative to vehicle-treated control cells (100%). Dose–response curves were plotted, and IC50 values (concentration of drug resulting in 50% reduction in cell viability) were obtained using Prism GraphPad 4.
Analysis of protein expression and cleavage by Western blotting
Cells were harvested in whole cell lysis buffer containing 62.5 mM Tris (pH 6.8), 2% (w/v) SDS, 10% (v/v) glycerol, 0.00125% (w/v) bromophenol blue and 50 mM DTT. The samples were then sonicated briefly and denatured at 65°C for 10 min before the separation of proteins on a polyacrylamide gel and transfer to PVDF membrane. The PVDF transfers were probed with primary monoclonal antibodies generated against P-glycoprotein, BCRP, Bcl-2, pro-caspase-3, PARP or β-actin all purchased from Merck Biosciences (Nottingham, UK), followed by incubation with a horseradish peroxidase-conjugated anti-mouse antibody (Promega, Madison, WI, USA). Protein expression was visualised by enhanced chemiluminescence.
Determination of DNA content
Following treatment, cells were harvested by centrifugation at 800g for 10 min. Cell pellets were resuspended in 200 μl PBS and fixed by a drop-wise addition of 2 ml of ice-cold 70% (v/v) ethanol/PBS while gently vortexing. Following overnight fixation at −20°C, the cells were again centrifuged to remove the ethanol and resuspended in PBS supplemented with 0.5 mg/ml RNase A and 0.15 mg/ml propidium iodide (PI). Cells were incubated in the dark at 37°C for 30 min. The PI fluorescence was measured on a linear scale using a FACSCalibur flow cytometer (Becton–Dickinson, San Jose, CA, USA). Data collections (10,000 events per sample) were gated to exclude cell debris and cell aggregates. PI fluorescence was proportional to the amount of DNA present in each entity and therefore indicated the stage of the cell cycle. Cells in G0/G1 were diploid (2N DNA content), cells in the S phase had DNA contents between 2N and 4N, cells in G2/M were tetraploid (4N DNA content), while apoptotic cells were hypoploid and contained <2N DNA. All data were recorded and analysed using the CellQuest software (Becton–Dickinson, San Jose, CA, USA).
Statistical analysis
Results were presented as mean ± SEM. The statistical analysis of experimental data was performed using the computer program Prism GraphPad 4. P values were determined using a two-tailed Student’s paired t test. A value of P < 0.05 was considered to be significant.
Preliminary molecular docking study
A computational docking experiment was undertaken to predict the likelihood of PBOX-6 interacting with the P-glycoprotein substrate binding site. Using the Penzotti study [27] as starting point, a dataset of 176 compounds was assembled (76 non-substrates and 100 substrates of P-glycoprotein). The software system MOE (Molecular Operating Environment) (Chemical Computing Group, Montreal, Canada) was used to process and refine the initial P-glycoprotein X-ray structure (Protein Data Bank ID: 3G60) [28], and Pipeline Pilot (Accelrys Software Inc., San Diego, CA, USA) processed the molecular database and generated ROC (receiver operating characteristic) data. Utilising OMEGA (Version: 2.3.2) (Openeye Scientific Software, Santa Fe, NM, USA), 200 conformers of each of the drug molecules were generated and docked using version 2.2.3 of the docking engine FRED (Fast Rigid Exhaustive Docking) (Openeye Scientific Software) into the QZ59-RRR substrate binding site of the recently determined X-ray structure of mammalian P-glycoprotein (3G60). The default values were used, except for the generation of 5,000 pose hit-list. The final stage of docking a ligand is to score the pose or poses generated by the docking steps; therefore, the ability of the 8 structure-based scoring functions available in FRED to discriminate between substrates and non-substrates was determined.
Results
Expression of MDR-ABC transporters in HL60 cells
HL60-MDR1 and HL60-ABCG2 cells were confirmed by Western blot to express the multi-drug transporters, P-glycoprotein or BCRP, respectively. The parental strain of HL60 cells did not express either transporter (Fig. 2). Each cell line was screened regularly during the course of this study to ensure that the expression of the transporters was maintained.

HL60-MDR1 cells expressed P-glycoprotein, while HL60-ABCG2 cells expressed BCRP. Whole cell extracts from HL60-parental, HL60-MDR1 and HL60-ABCG2 cells were separated by SDS–PAGE and transferred to PVDF. Blots were probed with monoclonal anti-P-glycoprotein or anti-BCRP (BXP-21) antibodies followed by a HRP-conjugated anti-mouse secondary. Blots were also probed for β-actin as a loading control
PBOX compounds reduced MDR HL60 cell proliferation
HL60, HL60-MDR1 and HL60-ABCG2 cells were treated for 72 h with a range of concentrations of PBOXs, paclitaxel, vincristine or SN-38 and cell proliferation measured by AlamarBlue assay (Table 1a). Our three representative PBOXs, PBOX-6, PBOX-15 and PBOX-16, reduced cell proliferation with similar efficacy in all three cell lines regardless of transporter-expression status. IC50 values were approximately 2 μM for PBOX-6 and approximately 0.2 μM for PBOX-15 and PBOX-16. The HL60-MDR1 cells proved resistant to other MTAs paclitaxel and vincristine. In HL60 parental strain, the IC50 value for paclitaxel was as low as 2.8 nM, whilst in HL60-MDR1 cells, even concentrations as high as 10 μM failed to reduce cell proliferation. Higher concentrations of paclitaxel could not be tested due to problems with solubility. Likewise, vincristine displayed an IC50 value of 2.5 nM in parental HL60 cells, whereas in HL60-MDR1, a concentration of 0.68 μM was required to reduce the cell proliferation by 50%, indicating a resistance factor of 268 between the parental and the P-glycoprotein-expressing HL60-MDR1 cells. Topoisomerase I inhibitor SN-38 had an IC50 value of 5.7 nM in HL60 cells, whereas in BCRP-expressing HL60-ABCG2 cells, the IC50 was 0.11 μM, indicating that these cells are 20 times more resistant to SN-38 than parental HL60 cells. The concentrations of drugs used for the remaining experiments performed in HL60 cells were chosen to reflect the IC50 values obtained by this cell proliferation assay.
PBOX compounds induced G2/M arrest and apoptosis in MDR HL60 cells
Microtubule-targeting agents PBOX-6 (10 μM), -15 (1 μM) and -16 (1 μM), paclitaxel (500 nM) and vincristine (100 nM) caused statistically significant increases in the percentage of parental HL60 cells found in the G2/M phase (4N DNA content) of the cell cycle (Fig. 3a) characteristic of tubulin disruption followed by an increase in the amount of hypoploid cells indicative of apoptosis (Fig. 3c). In HL60-MDR1 cells, although paclitaxel and vincristine failed to induce G2/M arrest or apoptosis, the three PBOX compounds were capable of inducing significant amounts (Fig. 3a, c). This suggested that while P-glycoprotein-expressing HL60-MDR1 cells are resistant to common MTAs such as paclitaxel and vincristine, they remain sensitive to the three PBOX compounds. Topoisomerase I inhibitor SN-38 (25 nM) induced an increase in the percentage of HL60 and HL60-MDR1 cells found in the S phase (2N–4N DNA content) of the cell cycle (Fig. 3b) followed by an increase in the percentage of apoptotic cells (Fig. 3c) but did not induce a significant response in HL60-ABCG2 cells indicating that these BCRP-expressing cells were resistant to SN-38. The PBOX compounds induced significant amounts of G2/M arrest (Fig. 3a) and apoptosis (Fig. 3c) in HL60-ABCG2 cells.

PBOX compounds induced G2/M arrest and apoptosis in HL60-parental, paclitaxel- and vincristine-resistant HL60-MDR1 and SN-38-resistant HL60-ABCG2 cells. Parental HL60, HL60-MDR1 and HL60-ABCG2 cells were treated with vehicle (0.5% EtOH), PBOX-6 (10 μM), PBOX-15 (1 μM), PBOX-16 (1 μM), paclitaxel (500 nM), vincristine (100 nM) or SN-38 (25 nM) for 8 h to measure G2/M (a) and S (b) phases or 48 h to quantify apoptosis (c). Cells were fixed in ethanol, stained with propidium iodide and DNA content assessed by flow cytometry. Analysis of data was performed using CellQuest. Cells in the subG0/G1 phase (<2N DNA) were deemed apoptotic, cells with 2N–4N DNA content were classed as S-phase, while cells with 4N quantities of DNA were considered to be in the G2/M phase of the cell cycle. Values represent the mean ± SEM for three independent experiments (***P < 0.0005; **P < 0.005; *P < 0.05)
PBOX compounds induced cleavage of Bcl-2, pro-caspase-3 and PARP in MDR HL60 cells
To confirm that the hypoploid cells we observed during cell cycle analysis were actually apoptotic, we assessed the cleavage of three proteins that are associated with the apoptotic process, namely, anti-apoptotic protein Bcl-2, pro-caspase-3 and DNA repair enzyme PARP (Fig. 4). In line with our DNA content analysis results, we found that all six drugs induced cleavage of Bcl-2, pro-caspase-3 and PARP in parental HL60 cells (Fig. 4a). However, treatment of HL60-MDR1 cells with paclitaxel or vincristine failed to induce cleavage of these same proteins (Fig. 4b), while HL60-ABCG2 cells displayed resistance to SN-38-induced cleavage (Fig. 4c). In contrast, both resistant cell lines remained sensitive to the effects of the PBOX-induced cleavage of these apoptosis-associated proteins. Expression of β-actin was used as a loading control in all experiments.

PBOX compounds induced cleavage of Bcl-2, pro-caspase-3 and PARP in HL60-parental and -resistant cells. Parental HL60 (a), HL60-MDR1 (b) and HL60-ABCG2 (c) cells were treated with vehicle (0.5% EtOH) (Veh), 10 μM PBOX-6 (P6), 1 μM PBOX-15 (P15), 1 μM PBOX-16 (P16), 500 nM paclitaxel (Tx), 100 nM vincristine (Vc) or 25 nM SN-38 (SN) for 48 h. Protein cleavage was assessed by Western blot using monoclonal antibodies generated against Bcl-2, pro-caspase-3, PARP or loading control β-actin followed by a HRP-conjugated anti-mouse secondary antibody. Blots are representative of three independent experiments
Verapamil, a P-glycoprotein inhibitor and fumitremorgan C, a BCRP inhibitor, reversed resistance in HL60-MDR1 and HL60-ABCG2 cells, respectively
We next wished to confirm that the resistance observed in HL60-MDR1 cells to paclitaxel and vincristine was caused by P-glycoprotein-mediated drug efflux. Therefore, we treated these cells with paclitaxel or vincristine in the presence of verapamil (10 μM). Verapamil is a high affinity substrate and ATPase stimulator of P-glycoprotein and hence is a competitive inhibitor of other substrates. In the presence of this inhibitor, sensitivity to paclitaxel and vincristine was restored leading to statistically significant levels of G2/M arrest (Fig. 5a), hypoploid apoptotic cells (Fig. 5b) and cleavage of Bcl-2, pro-caspase-3 and PARP (Fig. 5e). Similarly, we confirmed that resistance to SN-38 in HL60-ABCG2 was due to BCRP-mediated efflux, by treating cells with SN-38 in the presence of a specific BCRP inhibitor, fumitremorgan C (5 μM). In the presence of fumitremorgan C, sensitivity to SN-38 was restored leading to statistically significant levels of S-phase arrest (Fig. 5c) followed by apoptosis (Fig. 5d) and cleavage of Bcl-2, pro-caspase-3 and PARP (Fig. 5f). These results were specific as fumitremorgan C failed to restore sensitivity of HL60-MDR to paclitaxel and vincristine (Fig. 5e), whilst verapamil failed to restore sensitivity of HL60-ABCG2 cells to SN-38 (Fig. 5f).

Pre-treatment with P-glycoprotein inhibitor, verapamil or BCRP inhibitor, fumitremorgan C reversed paclitaxel and vincristine or SN-38 mediated resistance, respectively. HL60-MDR1 and HL60-ABCG2 cells were pre-treated with 10 μM verapamil (VP) or 5 μM fumitremorgan C (FTC) for 1 h followed by addition of vehicle (0.5% EtOH) (Veh), 500 nM paclitaxel (Tx), 100 nM vincristine (Vc) or 25 nM SN-38 (SN) for a further 8 h to detect G2/M (a) and S (c) phases or 48 h for apoptosis (b, d). The cells were fixed in ethanol, stained with propidium iodide and DNA content assessed by flow cytometry. Analysis of data was performed using the computer program CellQuest. Cells in the subG0/G1 phase (<2N DNA) were deemed apoptotic, cells with 2N–4N DNA content were classed as S-phase, while cells with 4N quantities of DNA were considered to be in the G2/M phase of the cell cycle. Values represent the mean ± SEM for three independent experiments (***P < 0.0005; **P < 0.005; *P < 0.05). Alternatively, whole cell lysates were prepared, proteins separated by SDS–PAGE and transferred to PVDF. Blots were then probed with monoclonal antibodies generated against Bcl-2, pro-caspase-3 or PARP followed by a HRP-conjugated anti-mouse secondary. Blots were also probed for β-actin as a loading control (e, f). Blots represent three independent experiments
Effect of PBOX compounds on A2780 and A2780-ADR cells
We also tested PBOX-6 and -16 in ovarian carcinoma A2780 parental cells and their adriamycin-resistant sub-strain A2780-ADR. We confirmed regularly during the course of these experiments that A2780-ADR cells expressed high levels of P-glycoprotein, while parental A2780 cells did not (Fig. 6a). We assessed the effect of PBOX-6 and -16, paclitaxel and vincristine on A2780 cell proliferation. The IC50 values were approximately 4 μM for PBOX-6 or between 0.2 and 0.7 μM for PBOX-16 in both parental and resistant A2780 cells, whereas A2780-ADR cells were more resistant to paclitaxel (greater than 1,946-fold) and vincristine (633-fold) than parental A2780 cells (Table 1b). These results indicated that like HL60-MDR1 cells, A2780-ADR displayed resistance to paclitaxel and vincristine but not to PBOX-6 and PBOX-16. Similarly, while treatment of parental A2780 cells with PBOXs, paclitaxel or vincristine lead to significant levels of G2/M arrest (Fig. 6b), A2780-ADR cells appeared resistant to paclitaxel- and vincristine-induced G2/M arrest (Fig. 6c). However, PBOX-6 and -16 were capable of inducing significant levels of G2/M arrest in these cells (Fig. 6c). Resistance to paclitaxel and vincristine was reversed by the presence of verapamil (Fig. 6d), confirming that resistance was due to the expression of P-glycoprotein rather than by other mechanisms of MDR.

P-glycoprotein-expressing ovarian carcinoma A2780-ADR cells are resistant to paclitaxel and vincristine but not PBOX compounds. Whole cell lysates from parental and adriamycin-resistant A2780 cells were prepared, proteins separated by SDS–PAGE and transferred to PVDF. Blots were then probed with a monoclonal anti-P-glycoprotein antibody followed by a HRP-conjugated anti-mouse secondary. Blots were also probed for β-actin as a loading control (a). Cells were treated with vehicle (0.5% EtOH), PBOX-6 (10 μM), PBOX-16 (1 μM), paclitaxel (1 μM) or vincristine (1 μM) for 24 h in the absence (b, c) or presence (d) of 10 μM verapamil (1 h pre-treatment). Cells were fixed in ethanol, stained with propidium iodide and DNA content assessed by flow cytometry. Analysis of data was performed using CellQuest. Cells with 4N quantities of DNA were considered to be in the G2/M phase of the cell cycle. Values represent the mean ± SEM for three independent experiments (***P < 0.0005; **P < 0.005)
Preliminary molecular docking of PBOX-6 in the P-glycoprotein substrate binding site
We performed a virtual molecular docking study to assess whether based on its structure, PBOX-6 was likely to be a substrate or non-substrate of the P-glycoprotein substrate binding site. Using molecular modelling software, conformers were generated and docked into the QZ59-RRR substrate binding site of P-glycoprotein. Finally, we scored the pose or poses generated by the docking steps. Chemgauss2, one of the eight scoring functions available in the docking engine FRED, uses smooth Gaussian functions to represent the shape and chemistry of molecules. This scoring function was judged to have the best performance with a ROC score of 0.704, and on examination of the hit-list, PBOX-6 was ranked at position 90, while paclitaxel and vincristine were ranked higher at positions 10 and 38, respectively.
Discussion
We have previously established that pro-apoptotic PBOX compounds may prove to be useful in the treatment of cancer. We have identified these compounds as novel microtubule-targeting agents, since they potently inhibit the assembly of tubulin in vitro [1] and have been demonstrated to cause microtubule depolymerisation in breast carcinoma MCF-7 cells [1], cervical carcinoma HeLa cells [29], human T lymphoma Hut78 cells [30], ovarian carcinoma A2780 cells [8], CML K562 cells [8], primary chronic lymphocytic leukaemia (CLL) cells [7] and primary HUVEC cells [8]. These microtubule-targeting PBOXs induced G2/M arrest (which is closely correlated with microtubular disruption) followed by apoptosis in cancer cell lines derived from both haematological malignancies and breast carcinoma [1–6], while in vivo PBOX-6 inhibited the growth of an aggressive mouse mammary tumour model [9]. In addition, we have recently reported that PBOX-6 in combination with tyrosine kinase inhibitor STI-571 can enhance apoptosis in STI-resistant CML K562 cells [31]. However, it has not yet been established whether PBOX compounds can similarly induce apoptosis in drug-resistant cancers caused by the expression of members of the ABC family of drug efflux transporters. Therefore, in this study, we investigated whether PBOX compounds may be potential candidates for the treatment of MDR cancers by comparing the efficacy of PBOX compounds in ABC transporter-negative cancer cells to cancer cells specifically expressing either P-glycoprotein or breast cancer resistance protein (BCRP), two of the main ABC transporters involved in MDR.
First, we examined the effects of three representative pro-apoptotic PBOX compounds (PBOX-6, -15 and -16) on cell viability, cell cycle profiles and apoptosis in parental human promyelocytic leukaemia HL60 cells and their drug-resistant substrains expressing either P-glycoprotein (HL60-MDR1 cells) or BCRP (HL60-ABCG2 cells). We established that PBOX-6, -15 and -16 were capable of reducing cell viability by inducing G2/M arrest followed by apoptosis in HL60 parental, HL60-MDR1 and HL60-ABCG2 cells. This suggested that PBOXs were effective in the destruction of leukaemia cells regardless of MDR transporter expression.
Although we showed by Western blot that our cells highly expressed the desired transporters, we also wished to confirm that the transporters present were fully functional. Therefore, we treated the HL60-MDR1 cells with known P-glycoprotein substrates paclitaxel and vincristine [32] and found that these cells were resistant to both drugs. To eliminate the possibility that the resistance we observed to paclitaxel and vincristine may have been caused by another mechanism of MDR, we also treated the cells with these MTAs in the presence of a competitive inhibitor of P-glycoprotein, verapamil [33]. This inhibitor successfully restored the sensitivity of the cells to paclitaxel and vincristine, illustrating that the resistance we observed was due to drug efflux through the P-glycoprotein transporter. Similarly, we treated HL60-ABCG2 cells with a known substrate of BCRP, SN-38 [21] and found the cells displayed resistance to this drug indicating that BCRP expressed in our cells appeared to be functional. Again to confirm SN-38 resistance was not mediated by some other mechanism of MDR, we treated the HL60-ABCG2 cells in the presence of a specific high-affinity inhibitor of BCRP, the micotoxin fumitremorgin C [34, 35] which proved capable of restoring sensitivity to SN-38. In combination, these results indicated that while the two MDR HL60 strains displayed resistance to other anti-cancer agents compared to parental HL60s, PBOX compounds were capable of producing similar responses in all three cell lines in spite of ABC transporter expression.
To support our findings in leukaemia cells, we also examined the effects of PBOX-6 and -16 on tumour cells derived from a solid malignancy, human ovarian carcinoma A2780 cells and their adriamycin-resistant substrain A2780-ADR which expressed P-glycoprotein. Similarly, while the A2780-ADR cells displayed resistance to paclitaxel and vincristine compared to parental A2780 cells, they did not demonstrate resistance to the PBOX compounds. Again, we confirmed that the P-glycoprotein was functional in the A2780-ADR cells by demonstrating that paclitaxel- and vincristine-induced resistance was reversed in the presence of verapamil.
Together, our in vitro data indicated that PBOX compounds were not substrates for the P-glycoprotein or BCRP transporters. The 170-kDa transmembrane transporter P-glycoprotein displays the broadest substrate specificity of the ABC family, extruding an array of structurally unrelated compounds. Its substrates are preferentially hydrophobic compounds that can partition into the lipid bilayer and also tend to have planar aromatic domains and tertiary amino groups. Substrates range, in molecular mass, from 300 to ~4,000 Da [28, 36]. Like P-glycoprotein, the half transporter BCRP has wide substrate specificity. Once dimerised, it can transport hydrophobic molecules (both positively and negatively charged) and large anionic compounds including many different drugs and metabolites (such as cytotoxic drugs and their partially detoxified metabolites) along with toxins and carcinogens found in food products [20]. To complement our findings in vitro, we wished to establish by virtual modelling, whether based on their structures, the PBOXs were likely to be substrates or non-substrates of ABC transporters.
As the X-ray structure of mammalian P-glycoprotein has recently been published [28], we decided to perform a series of computer-based experiments to preliminarily examine the molecular docking of PBOX compounds in the P-glycoprotein substrate binding site. PBOX-6 was chosen as the representative PBOX compound used in this investigation as it has been extensively studied and contains the central scaffold core features which are synthetically modified to yield other members of the PBOX series (Fig. 1). We used the Penzotti study [27] to assemble a dataset of 176 compounds that were classified into two groups based on whether they were substrates (100 compounds) or non-substrates (76 compounds) of P-glycoprotein. Substrates were classified as being compounds that were reported to be transported by P-glycoprotein, this group also included compounds that are reported to cause over-expression of P-glycoprotein and contribute to MDR. Non-substrates were classified as being compounds that are not transported by P-glycoprotein, this group also included compounds that are reported to be bound but not transported by P-glycoprotein; such compounds can be classed as possible inhibitors of P-glycoprotein.
Following the processing and refinement of the P-glycoprotein X-ray structure and the generation of conformers, PBOX-6 was virtually docked into the QZ59-RRR substrate binding site of P-glycoprotein, and a 5,000 pose hit-list generated. The best scoring function had a ROC score of 0.704. P-glycoprotein substrates should be ranked at the top of the hit-list. Interestingly, PBOX-6 was only found to be ranked at position 90 on the hit-list, while P-glycoprotein substrates, paclitaxel and vincristine, are highly ranked at positions 10 and 38, respectively. This corresponded to our in vitro data whereby cell lines that expressed P-glycoprotein (HL60-MDR and A2780-ADR) were more resistant to paclitaxel (resistance factor: >1,946) than vincristine (resistance factor: 258 and 633, respectively) and also displayed no resistance to PBOXs when compared to their P-glycoprotein negative parental strains.
A number of caveats need to be considered when performing docking studies on P-glycoprotein. The closer the ROC score is to 1, the more confident we can be that the docking programs performed accurately in distinguishing between substrates and non-substrates. The ROC score of 0.704 is far from ideal as the P-glycoprotein binding site has a large volume leading to difficulty in computationally predicting accurate binding poses. The differentiation in the literature between substrate (can also be an inducer) and non-substrate (may be an inhibitor) is not ideal for evaluating the reliability of a virtual screen docking protocol. With these factors in mind, this docking study should be viewed as indicative rather than quantitative, but it does show that for the X-ray crystallographic conformation of the P-glycoprotein substrate binding site, the PBOX compounds do not have the ability to form very favourable binding interactions.
Limitations associated with the use of microtubule-targeting agents such as taxanes include dose-limiting haematopoietic toxicity and cumulative neurotoxicity. As we have demonstrated that the PBOXs do not appear to be substrates for drug efflux pumps, they may be able to bypass such pumps which are present in the choroid plexus to exclude potentially neurotoxic species from the CNS. We have previously demonstrated that the PBOXs, at antitumoural efficacious doses in vivo, were well tolerated with no major systemic toxicity as indicated by no loss of body weight of the animals [9]. Additional data indicated that the PBOXs exerted no haematological toxicities (unpublished observations). Although we have yet to determine directly the neurotoxic effects of the PBOXs in vivo, it is of interest to note that some novel tubulin-binding agents such as the epothilone derivative, patupilone and the novel synthetic compound indibulin (D-24851), which are both currently undergoing clinical trials, do not appear to be susceptible to P-glycoprotein and similar drug efflux pumps, and yet they exhibit minimal neurotoxicity [37, 38]. Indibulin has been shown to discriminate between highly post-translationally modified tubulin present in mature neuronal microtubules and less-modified tubulin present in immature neuronal or non-neuronal microtubules [39]. Indibulin appears to be specific for non-modified tubulin, whereas vincristine acts on either tubulin equally well. It will be important in future studies to determine directly any neurotoxic effects of the PBOXs in vivo, for example, by the use of a coordination test (rota-rod) or by measuring nerve conductance velocity following administration of the PBOXs. However, what is promising is that in a previous study using these tests as an index of neurotoxicity with a variety of tubulin-binding agents such as vincristine, colchicine and the novel drug indibulin, neurotoxicity was strongly correlated with loss of body weight of the animals [38]. The PBOXs, at anti-tumoural efficacious doses in vivo, exert no effect on body weight, suggesting that they may also exhibit limited neurotoxicity.
In conclusion, our results suggest that pro-apoptotic PBOXs can induce cytotoxicity in MDR cancer cells expressing either P-glycoprotein or BCRP with similar potency to ABC transporter-negative cancer cells. In addition, based on their structure, PBOXs do not appear to be substrates of P-glycoprotein. Therefore, pro-apoptotic PBOXs may be suitable candidates for the treatment of P-glycoprotein- and BCRP-associated MDR cancers.
References
Mulligan J, Greene LM, Cloonan S et al (2006) Identification of tubulin as the molecular target of pro-apoptotic pyrrolo-1, 5-benzoxazepines. Mol Pharmacol 70:60–70
Zisterer DM, Campiani G, Nacci V et al (2000) Pyrrolo-1, 5-benzoxazepines induce apoptosis in HL-60, Jurkat, and Hut-78 cells: a new class of apoptotic agents. J Pharmacol Exp Ther 293:48–59
Mc Gee MM, Campiani G, Ramunno A et al (2001) Pyrrolo-1, 5-benzoxazepines induce apoptosis in chronic myelogenous leukemia (CML) cells by bypassing the apoptotic suppressor bcr-abl. J Pharmacol Exp Ther 296:31–40
Mc Gee MM, Hyland E, Campiani G et al (2002) Caspase-3 is not essential for DNA fragmentation in MCF-7 cells during apoptosis induced by the pyrrolo-1, 5-benzoxazepine, PBOX-6. FEBS Lett 515:66–70
Mc Gee MM, Campiani G, Ramunno A et al (2002) Activation of the c-Jun N-terminal kinase (JNK) signaling pathway is essential during PBOX-6-induced apoptosis in chronic myelogenous leukemia (CML) cells. J Biol Chem 277:18383–18389
Mc Gee MM, Greene LM, Ledwidge S et al (2004) Selective induction of apoptosis by the pyrrolo-1, 5-benzoxazepine 7-[[dimethylcarbamoyl]oxy]-6-(2-naphthyl)pyrrolo-[2, 1-d] (1, 5)-benzoxazepine (PBOX-6) in Leukemia cells occurs via the c-Jun NH2-terminal kinase-dependent phosphorylation and inactivation of Bcl-2 and Bcl-XL. J Pharmacol Exp Ther 310:1084–1095
McElligott AM, Maginn EN, Greene LM et al (2009) The novel tubulin targeting agent, pyrrolo-1, 5-benzoxazepine-15, induces apoptosis in poor prognostic subgroups of Chronic Lymphocytic Leukemia. Cancer Res 69:8366–8375
Nathwani SM, Butler S, Meegan MJ et al (2010) Dual targeting of tumour cells and host endothelial cells by novel microtubule-targeting agents, pyrrolo-1, 5-benzoxazepines. Cancer Chemother Pharmacol 65:289–300
Greene LM, Fleeton M, Mulligan J et al (2005) The pyrrolo-1, 5-benzoxazepine, PBOX-6, inhibits the growth of breast cancer cells in vitro independent of estrogen receptor status, and inhibits breast tumour growth in vivo. Oncol Rep 5:1357–1363
Mashima T, Seimiya H, Chen Z et al (1998) Apoptosis resistance in tumour cells. Cytotechnology 27:293–308
Cowan KH, Batist G, Tulpule A et al (1986) Similar biochemical changes associated with multidrug resistance in human breast cancer cells and carcinogen-induced resistance to xenobiotics in rats. Proc Natl Acad Sci USA 83:9328–9332
Redmond SM, Joncourt F, Buser K et al (1991) Assessment of P-glycoprotein, gluthathione-based detoxifying enzymes and O6-alkylguanine-DNA alkyltransferase as potential indicators of constitutive drug resistance in human colorectal tumours. Cancer Res 51:2092–2097
Eijdems EW, de Haas M, Timmerman AJ et al (1995) Reduced topoisomerase II activity in multidrug-resistant human non-small cell lung cancer cell lines. Br J Cancer 71:40–47
Sarkadi B, Homolya L, Szakacs G et al (2006) Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defence system. Physiol Rev 86:1179–1236
Maliepaard M, Scheffer GL, Faneyte IF et al (2001) Subcellular localisation and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 61:3458–3464
Scheffer GL, Maliepaard M, Pijnenborg AC (2000) Breast cancer resistance protein is localized at the plasma membrane in mitoxantrone- and topotecan-resistant cell lines. Cancer Res 60:2589–2593
Juliano RL, Ling V (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 455:152–162
Zaman GJR, Flens MJ, van Leusden MR et al (1994) The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Nat Acad Sci 91:8822–8826
Doyle LA, Yang W, Abruzzo LV et al (1998) A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA 95:15665–15670
Sarkadi B, Ozvegy-Laczka C, Nemet K et al (2004) ABCG2—a transporter for all seasons. FEBS Lett 567:116–120
Nakatomi K, Yoshikawa M, Oka M (2001) Transport of 7-ethyl-10-hydroxycamptothecin (SN-38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem Biophys Res Commun 288:827–832
Schellens JH, Maliepaard M, Scheper RJ et al (2000) Transport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implications. Ann NY Acad Sci 922:188–194
Volk EL, Schneider E (2003) Wild-type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter. Cancer Res 63:5538–5543
Borowski E, Bontemps-Gracz MM, Piwkowska A (2005) Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumour cells. Acta Biochim Pol 52:609–627
Ozvegy-Laczka C, Hegedus T, Varady G et al (2004) High-affinity interaction of tyrosine kinase inhibitors with ABCG2 multidrug transporter. Mol Pharmacol 65:1485–1495
Campiani G, Nacci V, Fiorini I et al (1996) Synthesis, biological activity, and SARs of pyrrolobenzoxazepine derivatives, a new class of specific “peripheral-type” benzodiazepine receptor ligands. J Med Chem 39:3435–3450
Penzotti JE, Lamb ML, Evensen E et al (2002) A computational ensemble pharmacophore model for identifying substrates of P-glycoprotein. J Med Chem 45:1737–1740
Aller SG, Yu J, Ward A et al (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323:1718–1722
Greene LM, Campiani G, Lawler M et al (2008) BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1, 5-benzoxazepines. Mol Pharmacol 73:419–430
Verma NK, Dempsey E, Conroy J et al (2008) A new microtubule-targeting compound PBOX-15 inhibits T-cell migration via post-translational modifications of tubulin. J Mol Med 86:457–469
Greene LM, Kelly L, Onnis V et al (2007) STI-571 enhances the apoptotic efficacy of PBOX-6, a novel microtubule targeting agent, in both STI-571-sensitive and -resistant Bcr-Abl-positive human chronic myeloid leukemia cells. J Pharmacol Exp Ther 321:288–297
Mollinedo F, Gajate C (2003) Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 8:413–450
Toffoli G, Simone F, Corona G et al (1995) Structure activity relationship of verapamil analogues and reversal of multidrug resistance. Biochem Pharmacol 50:1245–1255
Rabindran SK, He H, Singh M et al (1998) Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgan C. Cancer Res 58:5850–5858
Rabindran SK, Ross DD, Doyle LA et al (2000) Fumitremorgan C reverses drug resistance in cells transfected with breast cancer resistance protein. Cancer Res 60:47–50
Seelig A, Landwojtowicz E (2000) Structure-activity relationship of P-glycoprotein substrates and modifiers. Eur J Pharm Sci 12:31–40
Morris PG, Fornier MN (2008) Microtubule active agents: beyond the frontier. Clin Cancer Res 14:7167–7172
Bacher G, Nickel B, Emig P et al (2001) D-24851, a novel synthetic microtubule inhibitor, exerts curative antitumoral activity in vivo, shows efficacy toward multidrug-resistant tumour cells, and lacks neurotoxicity. Cancer Res 61:392–399
Wienecke A, Bacher G (2009) Indibulin, a novel microtubule inhibitor, discriminates between mature neuronal and nonneuronal tubulin. Cancer Res 69:171–177
Acknowledgments
This project was kindly funded by Science Foundation Ireland. In addition, the Molecular Design Group gratefully acknowledges the generous academic support afforded by the Chemical Computing Group (MOE software), OpenEye Scientific (OpenEye product suite), The Trinity Centre for High Performance Computing and Accelrys (Pipeline Pilot) in the delivery of this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nathwani, SM., Butler, S., Fayne, D. et al. Novel microtubule-targeting agents, pyrrolo-1,5-benzoxazepines, induce apoptosis in multi-drug-resistant cancer cells. Cancer Chemother Pharmacol 66, 585–596 (2010). https://doi.org/10.1007/s00280-009-1200-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-1200-9