
Abstract
Purpose
H1650 non-small cell lung cancer (NSCLC) cells display primary resistance to epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) although they have a deletion mutation on exon 19 of the EGFR gene. We investigated the effect of inhibition of both insulin-like growth factor receptor (IGFR) and EGFR signaling considering that IGFR signaling pathway has been implicated in the development and progression with therapeutic resistance of various cancers including lung cancer.
Methods
Three human NSCLC cell lines with an EGFR mutation of PC-9, HCC827 and H1650 were used for experiment. Cell viability and proliferative activity were assessed by MTT and three-dimensional culture assay. Combination index was obtained by CalcuSyn software. The change of EGFR- and IGFR-related signals was evaluated by western blots.
Results
H1650 cells were 1,000 times more resistant to gefitinib and erlotinib than HCC827 and PC-9 cells possessing the same EGFR mutation. Phosphatase and tensin homolog loss and sustained phosphorylation of Akt in spite of treatment with gefitinib were evident only in H1650 cells. Interestingly, IGFR phosphorylation was decreased by gefitinib in HCC827 and PC-9 cells while being maintained in H1650 cells. Combined treatment with the IGFR inhibitors α-IR3 and AG1024 enhanced gefitinib-induced growth inhibition and apoptosis, and down-regulated phosphorylation of Akt, EGFR and IGFR.
Conclusion
Combined inhibition of IGFR signaling enhances the growth inhibitory and apoptosis-inducing effects of gefitinib, suggesting that this approach could be useful to overcome the primary resistance to EGFR-TKIs in lung cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Clinical benefits of epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) such as gefitinib and erlotinib have been demonstrated in non-small cell lung cancer (NSCLC) [1, 2]. These drugs can sometimes produce a rapid and dramatic improvement in patients with disseminated cancer [3]. However, their effect is not universal among patients, and clinical trials showing that they are more effective in certain groups of lung cancer patients such as females, those who have never smoked and those with certain adenocarcinoma histology led to the discovery of activating EGFR mutations on the TK domain [4–6]. The response to EGFR-TKIs is closely related with these mutations and EGFR mutations play an important role in the pathogenesis of cancer in the lungs of “never smokers” [5].
Although more than 20 EGFR mutations are known, clinical significance has been demonstrated in only a few such as G719A on exon 18, a deletion mutation on exon 19 and L858R on exon 21 [3, 4]. The deletion on exon 19 is one of the most prevalent EGFR mutations, and is probably the best indicator of response to EGFR-TKIs [7]. The response rate in patients with EGFR mutations is approximately 70% [7]. It is increased if mutations without proven clinical relevance are excluded, and even more when only deletion mutations are considered. Nevertheless, it is quite certain that there are patients with activating EGFR mutations who show primary resistance. This is supported by the presence of H1650, a lung cancer cell line with deletion mutation on exon 19, which displays resistance although it does not exhibit known resistant mechanisms including T790M [8].
The insulin-like growth factor receptor (IGFR) signaling pathway has been implicated in the development and progression of various cancers including lung cancer [9, 10]. IGFR signaling can activate downstream signaling pathways such as extracellular signal-regulated protein kinase/mitogen-activated protein kinase (ERK/MAPK) and phosphoinositide 3-kinase (PI3K)/Akt, which are also modulated by EGFR [11, 12]. This raises the possibility that IGFR signaling is involved in resistance to EGFR-TKIs such as mesenchymal–epithelial transition factor (MET) signaling, which stimulates Akt activation to compensate for block of EGFR function by EGFR-TKIs [13]. In addition, similar resistance is also induced by IGFR signals in primary human glioblastoma, colorectal cancer and breast cancer cells [14, 15].
We investigated whether IGFR signaling pathway is involved in primary resistance to EGFR-TKIs and so would represent a reasonable target to overcome resistance using three NSCLC lines possessing the same deletion mutation on exon 19.
Materials and methods
Cell culture and reagents
The human NSCLC cell lines HCC827 and H1650 were obtained from the American Type Culture Collection (Rockville, MD, USA). PC-9 NSCLC cells were a kind gift from F. Koizumi and K. Nishio (National Cancer Center Hospital, Tokyo, Japan). All cell lines were cultured in RPMI 1640 medium that contained 10% fetal bovine serum (FBS), 2 mM l-glutamine and 100 units/ml penicillin and streptomycin and were maintained at 37°C in a humidified chamber containing 5% CO2. Gefitinib and ZD6474 were kindly provided by AstraZeneca Korea (Seoul, Korea), and erlotinib was supplied by Roche Korea (Seoul, Korea). CL-387,785 and α-IR3 were purchased from Calbiochem (La Jolla, CA) and Abcam (Cambridge, MA, USA), respectively.
Cell proliferation assay and combination index (CI) analysis
Cell proliferation was measured by a colorimetric assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Cells (0.5 × 104 per well) were exposed to varying concentrations of gefitinib, erlotinib, ZD6474 and CL-387,785 in 96-well sterile plastic plates (Costar, New York, USA). After 72 h, 15 μl of MTT solution (5 mg/ml) was added to each well and incubated for 4 h. Then, crystalline formazan was solubilized with 10% (w/v) sodium dodecyl sulfate (SDS). The absorbance at 595 nm was read spectrophotometrically using a microplate reader. Combination effect was evaluated in H1650 cells by the MTT assay at a 1:1 ratio of gefitinib (μM) and α-IR3 (μg/ml), and a 1:2 ratio of gefitinib (μM) and AG1024 (μM). The fraction affected (Fa) (i.e., Fa of 0.25 is equivalent to 75% viable cells) and CI values were processed by the CalcuSyn software (Biosoft, Cambridge, UK). CI values <1, 1 and >1 indicated synergism, additive effect and antagonism, respectively.
Apoptosis assay
Apoptotic cell death was determined by the Annexin V-fluorescein isothiocyanate (FITC) apoptosis kit I (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Briefly, cells were harvested, washed with phosphate-buffered saline, and resuspended in 1× binding buffer [10 mM Hepes (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2]. Cells were stained with 5 μl of annexin V-FITC and 10 μl of propidium iodide (PI), and incubated for 12 min at room temperature in the dark. 400 μl of 1× binding buffer was added and the samples were analyzed with Becton–Dickinson FACScan flow cytometer using CellQuest Pro software (BD Biosciences).
Three-dimensional culture assay
Wells in 24-well plates were pre-coated with 200 μl of growth factor reduced (GFR) matrigel (BD Biosciences) and placed in a 37°C incubator for 30 min for matrigel solidification. H1650 cells (2 × 104 per well) were incubated in the presence or absence of 1 μM gefitinib and 1 μg/ml α-IR3 for 72 h in the matrigel-coated wells. As the drugs were exhausted from the medium over time, fresh drug-supplemented medium was added every 3 days over 10 days. Cells were trypsinized and cell numbers were determined with an ADAM-MC automatic cell counter (NanoEnTek, Seoul, Korea) according to the manufacturer’s instructions.
Western blot analysis
Cellular lysates were prepared by suspending the cells in 100 μl of lysis buffer (137 mM NaCl, 15 mM EGTA, 0.1 mM sodium orthovanadate, 15 mM MgCl2, 0.1% Triton X-100, 25 mM MOPS, 100 μM phenylmethylsulfonyl fluoride, and 20 μM leupeptin, adjusted to pH 7.2). The proteins were separated on SDS–polyacrylamide gels and were electrotransferred to Immobilon-P membranes (Millipore, Bedford, MA, USA). Antibodies were obtained from the following sources: p-EGFR (Tyr1173), EGFR, IGF-1Rβ, MET, p-Akt (Ser 473), Akt and Erk were obtained form Santa Cruz Biotechnology (Santa Cruz, CA); p-HER2 (Tyr1221/22), p-ErbB3 (Tyr1222), p-MET (Tyr1234/1235), p-IGF-1R (Tyr1135/1136), p-Erk (Thr202/Tyr204), PTEN, caspase-3, caspase-8 and caspase-9 were purchased from Cell Signaling Technology (Beverly, MA); and β-actin was obtained from Sigma–Aldrich (St. Louis, MO, USA). Detection of specific proteins was carried out with an enhanced chemiluminescence Western blotting kit (Amersham Biosciences, NJ, USA) according to the manufacturer’s instructions. The ratio of phosphorylated/total IGFR expression was compared using densitometry by NIH Image J software.
Fluorescence in situ hybridization (FISH) for MET amplification
Fluorescence in situ hybridization was performed using a MET/CEP7 probe according to the manufacturer’s protocol (Abbott Molecular, Abbott Park, IL, USA). Briefly, the fixed slides were pretreated for 3 h in 2× NaCl-sodium citrate buffer (SSC) at 75°C, digested with proteinase K at 37°C for 60 min, codenatured at 85°C for 15 min, hybridized for approximately 36 h and washed with 2× SCC containing 0.3% NP-40. Chromatin was counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Abbott Molecular). Analysis was performed using an epifluorescence microscope with a single interference filter set for green (FITC), red (Texas red) and blue (DAPI), as well as dual (red/green) and triple (blue, red, green) band pass filters.
Results
Primary resistance of H1650 cells to EGFR-TKIs
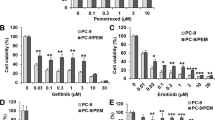
The effects of EGFR-TKIs on cell viability were determined by the MTT assay in HCC827, PC-9 and H1650 cells, which have the same deletion mutation on exon 19 of the EGFR gene (delE746-A750). Cells were treated with various concentrations of gefitinib, erlotinib, ZD6474 (an EGFR-VEGFR dual inhibitor) and CL-387,785 (an irreversible EGFR-TK inhibitor) for 72 h. IC50 values of EGFR-TKIs are summarized in Table 1. H1650 cells showed more than 1,000-fold higher IC50 for gefitinib than HCC827 and PC-9 cells. In addition, H1650 cells were also more resistant to erlotinib, ZD6474 and CL-387,785, suggesting that the presence of EGFR mutation was not sufficient to predict the sensitivity of EGFR-TKIs.
The apoptosis-inducing effect of gefitinib was evaluated by FACScan and western blot analyses. Apoptosis occurred in HCC827 and PC-9 cells treated with 0.01 μM gefitinib for 24 h, while concentrations of up to 1 μM could not induce apoptosis in H1650 cells, consistent with MTT assay results (Fig. 1a). Under identical conditions of apoptosis analysis, gefitinib activated caspase-3, -8 and -9, and cleaved poly(ADP-ribose) polymerase-1 (PARP-1), an intracellular substrate of caspase-3, in HCC827 and PC-9 cells (Fig. 1b, c). However, these results were not evident in H1650 cells, demonstrating the primary resistance to EGFR-TKIs.

Differential effects of gefitinib on apoptosis and EGFR-related signals. a HCC827, PC-9 and H1650 cells were treated with the indicated doses of gefitinib for 24 h in a medium containing 1% FBS. Cells were harvested, stained with Annexin V-FITC and PI, and analyzed by flow cytometry. The percentage of apoptotic cells was expressed as the sum of the bottom right (early state of apoptosis) and top right quadrants (late stage of apoptosis). Results were obtained from three experiments, and the bars represent standard deviation (SD). b Under identical conditions of apoptotic analysis, EGFR and apoptosis-related molecules were analyzed using western blot for indicated antibodies after treatment with gefitinib of various concentrations. c The EGFR mutant cell lines were treated with 1 μM gefitinib for indicated times and protein (40 μg) of cell lysates was analyzed by western blot
Persistence of phospho-Akt and phospho-IGFR in gefitinib-treated H1650 cells
The effect of gefitinib on Akt and ERK phosphorylation in EGFR downstream signals was examined, considering that the Akt and ERK pathways are mainly involved in gefitinib sensitivity and therapeutic efficacy [16]. Although phosphorylation of EGFR and ERK was inhibited by gefitinib in all cell lines, activated Akt persistently remained only in H1650 cells indicating that other signaling pathways may exist to maintain it. Interestingly, we also found that the treatment of gefitinib for 48 h modulated IGFR phosphorylation in HCC827 and PC-9 cells, but not in H1650 cells (Fig. 1b, c). These results are consistent with the suggestion that IGFR signaling could contribute to sustained Akt activity leading to the primary resistance to EGFR-TKIs in H1650.
Lack of resistance mechanisms such as T790M mutation and MET gene amplification in H1650 cells
The expression of baseline EGFR-related signaling molecules was explored by western blots in the three cell lines. Although results varied markedly in the cell lines, loss of phosphatase and tensin homolog (PTEN), which acts as a negative regulator of Akt and elevated expression of phospho-IGFR in H1650 were distinguished regarding resistance to EGFR-TKIs (Fig. 2a). The densitometric analysis showed that the phosphorylated/total IGFR ratio of HCC827, PC-9 and H1650 was 0.26, 0.43 and 1.73, respectively. No known resistant mechanisms such as T790M mutation leading to reduced efficacy of EGFR-TKIs were evident (data not shown). The MET/CEP7 ratio of HCC827, PC-9 and H1650 was 0.64, 0.67 and 0.72, respectively, indicating that the MET gene was not amplified in all cell lines (Fig. 2b).

Comparison of baseline EGFR-related signals having possible roles in resistance and MET amplification. a HCC827, PC-9 and H1650 cells were incubated in 10% FBS medium. Equal amounts of whole cell lysates (40 μg) were subjected to electrophoresis and the proteins were analyzed by western blot for indicated antibodies. b MET amplification was detected by FISH using probe against centromere of chromosome 7 (CEP7) and MET (×400)
AG1024 enhanced growth inhibitory and apoptotic effects of gefitinib in H1650 cells
To examine the growth inhibitory effect of combined inhibition of EGFR and IGFR signals, we treated with gefitinib and AG1024 (a small molecule that inhibits IGFR-TK) for 72 h at a concentration ratio of 1:2. Fa and CI values were 0.57 and 0.19, respectively, upon treatment with 5 μM gefitinib and 10 μM AG1024, indicative of a synergistic effect (Fig. 3a, b). A similar synergistic effect was observed over the entire range of tested concentrations. FACScan apoptosis analysis showed that AG1024 also enhanced the apoptosis-inducing effect of gefitinib (Fig. 3c). Consistent with this, caspase-3, -8 and -9 were activated and PARP-1 cleavage occurred with the combined treatment, as detected by western blots (Fig. 3d).

Enhanced growth inhibitory and apoptotic effects by combination with AG1024 and gefitinib. H1650 cells were cultured in medium containing 1% FBS and treated with gefitinib, AG1024 and their combinations for 72 h. Combined effects at a constant ratio (1:2 μM) were measured by a MTT assay. Dose–effect curve (a) and CI plots (b) were computationally generated; CI <1, 1 and >1 indicates synergism, additive effect and antagonism, respectively. c H1650 cells were treated with AG1024 and gefitinib for 48 h in a medium containing 1% FBS. Cells were harvested, stained with Annexin V-FITC and PI, and analyzed by flow cytometry. The percentage of apoptotic cells was expressed as the sum of the bottom right (the early state of apoptosis) and top right quadrants (the late stage of apoptosis). Results were obtained from three experiments, and the bars represent SD. *P < 0.01, **P < 0.005 vs. control and either drug alone. d Whole cell lysates (40 μg) were analyzed by western blot for caspase-8, caspase-9, caspase-3, PARP-1 and β-actin
Synergistic effects in combination with α-IR3, which suppresses Akt phosphorylation
We investigated whether α-IR3, a monoclonal blocking antibody for IGFR, enhanced the effect of gefitinib. Fa and CI values were 0.34 and 0.09, respectively, upon treatment with 1 μM gefitinib and 1 μg/ml α-IR3, indicative of synergistic growth inhibition (Fig. 4a, b). To further confirm the cellular effects of this combination treatment, H1650 cells were treatment with gefitinib and α-IR3 for 72 h in matrigel-coated wells. After elimination of drugs, cells were incubated for 10 days and then the viable cell numbers were ascertained. Colony formation was significantly reduced by the combination treatment compared with single drug treatment (Fig. 4c). Western blots clearly demonstrated that phosphorylation of Akt was completely suppressed by inhibition of both EGFR and IGFR signals (Fig. 4d), although H1650 cells do not possess PTEN activity.

Synergistic effects on growth inhibition by α-IR3 and gefitinib. H1650 cells were treated with 1 μM gefitinib, 1 μg/ml α-IR3 or a 1:1 ratio combination of both, and the effects were measured by a MTT assay. Dose–effect curve (a) and CI plots (b) were generated. c H1650 cells were incubated with 1 μM gefitinib and 1 μg/ml α-IR3 for 72 h in GFR matrigel-coated wells and then the culture medium was changed every 3 days within 10 days (phase contrast, ×20). Cells were trypsinized and cell numbers were determined. Bars represent the mean ± SD of three wells. d Under identical MTT assay conditions, cells were harvested and whole cell lysates (40 μg) were analyzed using western blot for indicated antibodies
Discussion
Examinations of clinical samples from patients who developed EGFR-TKI resistance during therapy and resistant cell lines generated by chronic exposure to drugs have identified two major mechanisms of resistance acquisition: T790M mutation and MET amplification [13, 17]. These resistance mechanisms seem to be of primary importance, considering their presence in specimens from EGFR-TKI-naïve patients [18, 19]. Moreover, H1975 lung cancer cells harboring T790M mutation show resistance to EGFR-TKIs despite the presence of a L858R activating mutant [17]. However, caution is needed in interpreting T790M mutation analysis because conventional direct sequencing could easily miss the presence of T790M, which requires more than 20% of target DNA for detection [18, 20]. In allelic dilution theory, small proportions of T790M among total tumor DNA can be related with resistance [21]. Therefore, we performed the T790M mutation test using the more sensitive Scorpion method than direct sequencing, which confirmed that resistance in H1650 cells was not caused by T790M mutation. In addition, MET amplification could not be detected by FISH analysis in HCC827, PC-9 and H1650 cells, showing that the two known resistant mechanisms, T790M mutation and MET gene amplification, are not correlated with primary resistance to EGFR-TKIs in H1650 cells.
EGFR-TKI sensitivity is largely dependent on Akt activation [22]. Persistently, phosphorylated Akt signal is a common characteristic in resistant cells, although its activity can be slightly reduced by EGFR-TKIs. This phosphorylated Akt tail may be caused by activation of EGFR families, PTEN loss and activation of other receptor-tyrosine kinase pathways, including c-MET and IGFR [8, 13, 23, 24]. H1650 cells express lower levels of phosphorylated and total EGFR, Her2 and Her3 and PTEN loss, which may lead to failure of Akt modulation and EGFR-TKI resistance. Although Her3 expression is required to activate the Akt pathway in gefitinib-sensitive NSCLC cell lines, introduction of Her3 protein fails to increase sensitivity in gefitinib-insensitive NSCLC cell lines [23]. Indeed, the already-low expression level of phosphorylated Her3 was not changed by gefitinib in H1650 cells suggesting that activation of Akt may not be maintained by Her3/Akt pathway (data not shown). PTEN loss leads to erlotinib resistance in H1650 cells [8]. Nevertheless, constitutive activation of Akt may play a more important role of erlotinib resistance than PTEN loss in EGFR mutant NSCLC cells [8]. Although we did not directly manipulate PTEN activity in H1650 cells, our results showing that combined inhibition of IGFR and EGFR could effectively suppress Akt activation suggest that its contribution might not be major.
Rather, our findings that gefitinib itself could inhibit IGFR activity in sensitive cells, but not in H1650 cells, indicate a possible role of IGFR signaling in primary resistance. IGFR signaling may play a role in maintaining Akt activation to compensate for gefitinib-mediated blockage of the EGFR pathway. Consistent with this, it has been demonstrated that gefitinib-resistant cells have hyperphosphorylation of the IGFR and constitutive association of IRS-1 with PI3K [24]. Presently, inhibition of IGFR signaling disrupted the association of IRS-1 with PI3K, restoring the capability of gefitinib to suppress PI3K/Akt signaling and cell growth. In addition, cross-talk between IGFR and EGFR has a specific role in inducing EGFR-TKIs resistance in lung cancer cell lines [25], although the study was not related with EGFR mutations.
In summary, we have clearly demonstrated that the combined inhibition of IGFR enhances the growth inhibitory and apoptotic effects of gefitinib in H1650 cells. Although more cell lines could not be examined for evaluation of clinical feasibility because H1650 is the sole NSCLC cell line showing primary resistance despite activating EGFR mutation, we suggest that combined inhibition of EGFR and IGFR may provide an effective therapeutic strategy to overcome primary resistance to EGFR-TKIs in lung cancer.
References
Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC (2003) Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 290:2149–2158
Perez-Soler R (2004) Phase II clinical trial data with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib (OSI-774) in non-small-cell lung cancer. Clin Lung Cancer 6(Suppl 1):S20–S23
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H (2004) EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 101:13306–13311
Park K, Goto K (2006) A review of the benefit-risk profile of gefitinib in Asian patients with advanced non-small-cell lung cancer. Curr Med Res Opin 22:561–573
Sequist LV, Bell DW, Lynch TJ, Haber DA (2007) Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol 25:587–595
Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, Girard L, Peyton M, Gazdar AF, Minna JD, Garraway LA, Kashkar H, Pao W, Meyerson M, Thomas RK (2009) PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of akt and EGFR. Cancer Res 69:3256–3261
Pollak MN, Schernhammer ES, Hankinson SE (2004) Insulin-like growth factors and neoplasia. Nat Rev Cancer 4:505–518
LeRoith D, Roberts CT Jr (2003) The insulin-like growth factor system and cancer. Cancer Lett 195:127–137
Petley T, Graff K, Jiang W, Yang H, Florini J (1999) Variation among cell types in the signaling pathways by which IGF-I stimulates specific cellular responses. Horm Metab Res 31:70–76
Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR (1997) The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem 272:6653–6662
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043
Chakravarti A, Loeffler JS, Dyson NJ (2002) Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res 62:200–207
Jones HE, Goddard L, Gee JM, Hiscox S, Rubini M, Barrow D, Knowlden JM, Williams S, Wakeling AE, Nicholson RI (2004) Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; iressa) in human breast and prostate cancer cells. Endocr Relat Cancer 11:793–814
Ono M, Hirata A, Kometani T, Miyagawa M, Ueda S, Kinoshita H, Fujii T, Kuwano M (2004) Sensitivity to gefitinib (iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther 3:465–472
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2:e73
Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, Suehisa H, Ouchida M, Aoe K, Aoe M, Kiura K, Shimizu N, Date H (2006) Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 66:7854–7858
Kubo T, Yamamoto H, Lockwood WW, Valencia I, Soh J, Peyton M, Jida M, Otani H, Fujii T, Ouchida M, Takigawa N, Kiura K, Shimizu K, Date H, Minna JD, Varella-Garcia M, Lam WL, Gazdar AF, Toyooka S (2009) MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer 124:1778–1784
Milbury CA, Li J, Makrigiorgos GM (2009) PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem 55:632–640
Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, Tracy S, Zhao X, Heymach JV, Johnson BE, Cantley LC, Janne PA (2006) Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest 116:2695–2706
Cappuzzo F, Magrini E, Ceresoli GL, Bartolini S, Rossi E, Ludovini V, Gregorc V, Ligorio C, Cancellieri A, Damiani S, Spreafico A, Paties CT, Lombardo L, Calandri C, Bellezza G, Tonato M, Crino L (2004) Akt phosphorylation and gefitinib efficacy in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 96:1133–1141
Engelman JA, Janne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC (2005) ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci USA 102:3788–3793
Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, Engelman JA (2008) Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest 118:2609–2619
Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY (2007) Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res 13:2795–2803
Acknowledgments
We thank Sun Ah Park for expert technical assistance.
Conflict of interest statement
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Choi, Y.J., Rho, J.K., Jeon, Bs. et al. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: a lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother Pharmacol 66, 381–388 (2010). https://doi.org/10.1007/s00280-009-1174-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-1174-7