
Abstract
Purpose
Recent studies have demonstrated that frequent, low-dose metronomic (MET) dosing of cytotoxic agents may not only be as efficient as conventional maximum tolerated dose (MTD) chemotherapy but also less toxic. In this study, we investigated the therapeutic effect and safety of MET chemotherapy using cyclophosphamide (CTX) in rats with chemically induced hepatocellular carcinoma (HCC).
Methods
Rats received weekly intraperitoneal (i.p.) injections of diethylnitrosamine during 16 weeks for induction of HCC. The rats were divided into three groups: MTD group received 40 mg/kg CTX i.p. injection on days 1, 3, and 5 of a 21-day cycle; Control and MET groups received saline and 20 mg/kg CTX i.p. injection twice a week, respectively. The growth-modulating effects and overall survival were compared between the groups. Anti-angiogenic effects were evaluated by a measurement of endothelial cell and VEGFR-2 expression.
Results
At 6 weeks of therapy, MTD and MET chemotherapy resulted in a significant reduction in tumor number and size compared with Control group. MET chemotherapy showed more prolonged survival than MTD chemotherapy and Control groups (P < 0.05). MET chemotherapy resulted in a significant decrease in both the micro-vessel density and endothelial proliferation index (P < 0.01). Furthermore, MET chemotherapy led to a greater decrease in VEGFR-2 expression at the mRNA and protein levels (P < 0.01).
Conclusions
MET scheduling not only exhibits anti-tumor and anti-angiogenic effects, but also prolongs survival without major toxicities in a rat model of HCC. Our results suggest that MET chemotherapy has a high therapeutic value and should be considered for future clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common type of malignancy and the third leading of cancer death worldwide [1]. Currently, surgical resection or liver transplantation is the best therapeutic option for HCC. However, only a minority of patients with HCC are eligible for the curative options, because of multifocal lesions and underlying liver cirrhosis. Recurrence remains high after radical surgery, and the 5-year overall survival rate has been reported to be only 40–50% among surgical patients [2]. Liver transplantation in HCC is limited to a subset of patients with small HCC, with the very strict selection criteria, and does not guarantee survival for patients with HCC exceeding the transplant criteria. Therefore, chemotherapy has been the most used palliative option of HCC treatment for a long time. The conventional dosing strategy for chemotherapy is based on short bursts of the maximum tolerated dose (MTD), which involves direct killing or inhibition of proliferating tumor cells [3, 4]. However, MTD chemotherapy causes undesirable side effects normally associated with conventional cytotoxic chemotherapy strategies. Thus, conventional chemotherapy for HCC has limited value in clinical practice, since there are no established cytotoxic regimens proven to be effective against this cancer. In order to improve the prognosis of HCC patients, there is a great need for the development of a chemotherapy strategy which is truly effective against HCC. Low-dose, continuous anti-angiogenic treatment, so-called metronomic (MET) chemotherapy, involves administrating a chemotherapeutic drug at a reduced dose compared with conventional treatment, but at regular, more frequent intervals with no long rest periods [5]. In contrast to conventional MTD chemotherapy, MET chemotherapy not only has a cytotoxic effect against tumor cells but also exerts an anti-angiogenic property toward tumor-associated endothelial cells [6–8], because it may significantly inhibit the development of drug resistance by targeting endothelial cells rather than cancer cells.
Angiogenesis has an important role in the progression and metastatic spread of tumors [9, 10]. Vascular endothelial growth factor (VEGF) is a key angiogenic factor, and its overexpression in tumor cells enhances tumor growth and metastasis in several animal models by inducing angiogenesis [11, 12]. Furthermore, previous studies have reported that VEGF and VEGFRs are highly expressed in HCC and activation of the VEGF–VEGFR pathway increases the tube formation and migration activity of endothelial cells, resulting in enhanced neovascularization [13–15].
Recently, preclinical studies have reported that MET chemotherapy results in marked regression of tumors via not only an antitumor cell effect but also an anti-angiogenesis effect, in a number of cancer models [16–18]. However, the effect of MET chemotherapy scheduling has not been tested in an experimental model of HCC. In fact, patients with HCC tend to experience more severe toxicities related to chemotherapy compared with patients with other cancers, because most of the HCC patients also have liver cirrhosis. Thus, there is a need for a chemotherapeutic regimen minimizing toxicity but having strong antitumor effects for patients with HCC. In this study, we investigated whether MET chemotherapy scheduling has therapeutic efficacy in terms of antitumor and anti-angiogenic properties and safety compared with MTD chemotherapy in a rat model of HCC. To address these issues, we examined the effects of MET chemotherapy on suppressing tumor growth and prolonging survival in comparison with MTD chemotherapy. Additionally, the anti-angiogenesis effect of MET chemotherapy by decreasing the expression of endothelial cells and VEGFR-2 was also studied.
Materials and methods
Experimental design
Sprague Dawley (SD) rats received intraperitoneal (i.p.) injections of diethylnitrosamine (DEN; Sigma Chemical Co, St. Louis, MO, USA) at 50 mg/kg body weight once a week for 16 weeks for induction of HCC [19, 20]. Two rats were killed at intervals of 2 weeks until the development of HCC. Therapy began 17 weeks after DEN-induced HCC and the rats were killed for experiments to confirm the therapeutic effect of treatment after 6 weeks in each group. The rats were randomly divided into three groups: the MTD chemotherapy group received a 40 mg/kg cyclophosphamide (CTX; Sigma Chemical Co, St. Louis, MO, USA) i.p. injection on days 1, 3, and 5 of a 21-day cycle; the untreated control group (control) received an i.p. injection of saline twice a week; and the MET chemotherapy group received a 20 mg/kg CTX i.p. injection twice a week. Animal care and the experiments followed the guidelines for the Care and Use of Laboratory Animals from the Research Supporting Center for Medical Science of The Catholic University of Korea.
Macroscopic and microscopic observations
The whole liver tissues were sliced into 2 mm thicknesses. Nodules of the liver were counted by macroscopic examination of the liver through two independent investigators according to the following criteria: nodules with dyschromatic and dysmorphic patterns comprised the N1 (3 mm ≤ x < 5 mm in diameter), N2 (5 mm ≤ x < 10 mm in diameter), and N3 groups (x ≥ 10 mm in diameter). Liver tissue samples were fixed in 10% formalin for 24 h before being embedded in paraffin. The paraffin-embedded sections were cut into 3 μm thicknesses, dewaxed, dehydrated, and stained with hematoxylin and eosin (H&E).
Immunohistochemical analysis
For immunohistochemical staining of proliferating cell nuclear antigen (PCNA) and von-Willebrand factor (vWF), the paraffin sections were dewaxed in xylene and rehydrated through a graded alcohol series. To block endogenous peroxidase activity, the sections were quenched in 3% hydrogen peroxidase. Next, the slides were blocked with normal serum blocking solution (DakoCytomation, Glostrup, Denmark), followed by incubation with primary antibody against PCNA and vWF (Abcam, Cambridge, UK) at 4°C for overnight. After rinsing the slides, the secondary antibody conjugated in a biotinylated link universal solution (Dakocytomation, Glostrup, Denmark) was applied. Then, the streptavidin-peroxidase solution (Dakocytomation, Glostrup, Denmark) was applied and the slides were incubated for 1 h at room temperature. Horseradish peroxidase (HRP) was detected with 3,3-diaminobenzidine (DAB; Vector Laboratories, Burlingame, CA, USA), and counterstained with hematoxylin. Stained sections were counted in five high-density fields at ×400 magnifications, and the mean endothelial cells density was recorded.
Detection of apoptotic cells in tumor tissues
Liver tissue samples were fixed in 10% formalin before being embedded in paraffin. The paraffin-embedded sections were cut into 3 μm thicknesses. Each section was dewaxed and rehydrated. Apoptotic cells were determined using the “Terminal-Deoxynucleotidyl-Transferase-mediated dUTP-Nick End Labeling” method as described previously [21], with the “ApopDETEK kit” (DakoCytomation, Glostrup, Denmark). In a final step, slices were counterstained with 4′-6-diamidino-2-phenylindole (DAPI), and examined by fluorescence microscopy. The total number of apoptotic cells in five randomly selected fields was counted at ×200 magnification. The apoptosis index was calculated as the percentage of positive-staining cells (apoptosis index = number of apoptotic cells/total number of nucleated cells × 100).
Total RNA extraction and semi-quantitative RT-PCR
Total RNA was extracted from frozen tissues using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. RNAs were reverse transcribed to single-strand cDNAs using random primer (TaKaRa Bio, Shiga, Japan) with SuperScript First Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). 2 μg of total RNA was amplified by polymerase chain reaction (PCR), and the cDNAs were amplified. The primer sequences were as follows: sense, 5′-ACT GCG CTC ATC TCC TGT TT-3′ and antisense, 5′-TTG CGT CTG AGG TCC TTC TT-3′ for VEGFR-2 (166 base pairs); and sense, 5′-AGA CAG CCG CAT CTT CTT GT-3′ and antisense, 5′-CTT GCC GTG GGT AGA GTC AT-3′ for GAPDH (207 base pairs). PCR conditions were carried out with an initial denaturation of 94°C for 5 min, followed by 30–35 cycles of denaturation at 94°C for 1 min, annealing at 54°C for 1 min, and extension at 72°C for 1 min. A final extension for 5 min at 72°C was carried out. PCR fragments were analyzed on 1.5% agarose gels stained with ethidium bromide.
Immunoblot analysis
The tissues were homogenized in t-per tissue protein extraction buffer (Pierce, Rockford, IL, USA). The lysates were cleared by centrifugation for 30 min at 4°C, and the supernatant kept frozen at −70°C. Proteins (40 μg) were separated by 8% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membranes (Whatman, Maidstone, Kent, UK). Then, membranes were blocked with 5% skim milk in PBS at room temperature for 30 min. The membranes were incubated with monoclonal anti-β-actin (Sigma-Aldrich, St. Louis, MO, USA), PCNA (Abcam, Cambridge, UK), and anti-VEGFR-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C for overnight. Each membrane was washed three times with 0.05% Tween-20 containing TBS (TBS-T), following incubated with horseradish peroxidase-conjugated anti-mouse secondary antibody (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Then, the specific protein bands were visualized with Enhanced Chemiluminescent system (ECL; Amersham, Biosciences, Little Chalfont, Buckinghamshire, UK) according to the manufacturer’s instructions. The relative intensity of the signals was analyzed using the Luminescent Image Analyzer LAS-4000 Plus and Image software Gauge 4.0 (FUJI Photo Film, Co. Ltd, Japan).
Statistics
Continuous variables were compared with the t test (between two groups) or ANOVA (for all groups) if normally distributed, and the Mann–Whitney rank-sum test or Kruskal–Wallis test (for all groups) if nonparametric. Survival curves were plotted by the method of Kaplan and Meier and tested for survival differences with the log-rank statistic. P values of less than 0.05 were used for statistical significance. All statistical analyses were performed using SPSS software (version 13.0, SPSS INC., Chicago, IL, USA).
Results
Sequential development of cirrhosis and HCC during the administration of DEN
During the planned DEN administration (50 mg/kg/week), periportal fibrosis with occasional bridging fibrosis was confirmed at microscopic examination of the liver at 10 weeks. From 12 weeks of therapy, microscopic fibrosis became obvious and distinct cirrhosis appeared at 14 weeks (data not shown). Malignant nodules with a diameter ≥3 mm and a dysmorphic/dyschromic aspect were detected on the surface of the liver after 16 weeks of DEN administration (Fig. 1a, b), and ultimately identified as well-differentiated HCC on histological examination (Fig. 1c, d).

DEN-induced HCC. a and b Gross findings of rat liver in DEN-induced HCC, respectively. Rat liver showed multinodular HCC after 16 weeks. c and d Pathological findings of DEN-induced HCC in a rat model with hematoxylin and eosin (H&E) staining (magnification ×40, × 200, respectively)
Assessment of toxicity
The median survival time of the rats treated with MTD chemotherapy at 100, 80, and 60 mg/kg CTX doses was 6.0 ± 0.2, 8.0 ± 0.0, and 11.0 ± 7.8 days, respectively. The reason for the early death of rats treated with MTD therapy appeared to be the cytotoxic effect of MTD chemotherapy (data not shown). Rat body weight loss was used as a parameter for toxicity; the rats were weighed once per week to determine any toxic effect of the drug. The body weights of the rats that received MTD chemotherapy and MET chemotherapy were 484.7 ± 79.3 and 498.8 ± 41.4 g, respectively, and were not significantly different after 6 weeks of treatment. However, the body weight of the MTD chemotherapy group tended to be lower than that of the control group (517.1 ± 44.6 g) (P = 0.133).
Prolonged survival with MET chemotherapy
The treatment of rats began at 16 weeks after confirmation of HCC and continued until the rats were killed. We analyzed the effects of MET chemotherapy on survival in the rat model of HCC. We found that the median survival time of the MET chemotherapy group was 46.0 ± 4.5 days, which was significantly better than the control group (26.0 ± 2.1 days) or the MTD chemotherapy group (28.0 ± 9.0 days) (log-rank test: P = 0.001 and P = 0.014, respectively). In contrast, the MTD chemotherapy group had no significant survival benefit when compared with the control group (P = 0.356) (Fig. 2).

Survival of rats according to different treatment strategies (n = 13–17 per group). Treatment was continued until the rats were individually moribund, and the days of life were recorded. Survival time of the MET chemotherapy group was significantly longer than the control and the MTD chemotherapy groups (log-rank test: P = 0.001 and P = 0.014, respectively)
Effect of MET chemotherapy on reducing the growth and number of tumors
The effect of treatment on tumor growth suppression was assessed by the measurement of the size and number of tumors at 6 weeks of treatment. As shown in Fig. 3a, an increase in both tumor number and size was observed in the control group compared with the MTD or MET chemotherapy groups. Furthermore, on the assessment of the liver and body weight, the control group revealed a significantly increased liver/body ratio than MTD or MET chemotherapy group, indicating a larger tumor burden in the control group (P = 0.029 and P = 0.003, respectively) (Fig. 3b). The size distribution of nodules was summarized in Table 1.

Effect of MET chemotherapy on inhibition of tumor growth. a Gross findings of DEN-treated rat livers after treatment for 6 weeks. b Percentage of liver/body weight ratio in rats after 6 weeks of therapy (*P < 0.05 and **P < 0.01)
Suppression of tumor cell proliferation by MET chemotherapy
To investigate the suppressive effect of MET chemotherapy on the proliferation of tumor cells, we performed two distinct assays. Immunohistochemical staining was performed on tumor sections from the above experiments and was stained with PCNA antibody, a cell proliferation marker. We confirmed that the MET and MTD chemotherapy groups had fewer PCNA-positive cells compared with the control group (Fig. 4a). We then showed that the MTD and MET chemotherapy groups had down-regulated expression of PCNA compared with the control group, as confirmed by an analysis of band densities (Fig. 4b). Both the MTD and MET chemotherapy groups showed a significantly decreased number of proliferating cells compared with the control group (both P < 0.01) (Fig. 4c).

Effect of MET chemotherapy on inhibition of tumor cell proliferation and induction of cell apoptosis. a Representative tumor sections prepared 6 weeks after treatment with saline (control), MTD, and MET. Tumor sections were stained against an anti-PCNA to detect proliferating cells (arrowheads) (magnification ×100). b Protein expression of PCNA after treatment for 6 weeks by immunoblot analysis. β-actin was used as a loading control. c Bar graph of the band intensities on PCNA expression. PCNA expression was quantified by densitometry with an Image software Gauge 4.0 program (**P < 0.01). d TUNEL-positive cells (arrowheads) in the tumor sections prepared after treatment with saline (control), MTD, and MET at 6 weeks (magnification ×200). e Bar graph of the mean percentage of TUNEL-positive cells. Five fields per slide, and at least three slides per group, were examined, and the statistical analysis was performed by ANOVA (*P < 0.05 and ***P < 0.001)
Induction of tumor cell apoptosis by MET chemotherapy
The sections were stained with the TUNEL agent and examined by fluorescence microscopy to confirm apoptotic tumor cells. The apoptotic cells in the sections were counted to record the apoptosis index. There were increased numbers of apoptotic cells in the MTD (21.9 ± 2.1%) and MET chemotherapy (17.8 ± 2.3%) groups compared with the control group (11.3 ± 1.9%) (Fig. 4d). Both the MTD and MET chemotherapy groups showed a significantly increased number of apoptotic cells compared with the control group (193.8 and 157.5%, respectively; both P < 0.001) (Fig. 4e).
Anti-angiogenic effect of MET chemotherapy
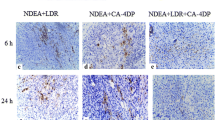
To evaluate the anti-angiogenic effect of the MET chemotherapy, liver tissues were sectioned and stained with an anti-vWF antibody to visualize micro-vessels. Tumor microvessel density (MVD) determined using vWF antibody in the chemotherapy groups was decreased compared with the control group. Moreover, the MET chemotherapy group had a greater decrease in endothelial cell expression than the MTD chemotherapy group (Fig. 5a). Overall, the MVD of the MET chemotherapy group was significantly reduced by 76.6 and 42.2%, respectively, compared with the control and MTD chemotherapy groups (Fig. 5b).

Effect of MET chemotherapy on inhibition of tumor angiogenesis. a vWF-positive endothelial cells (arrowheads) in the tumor sections prepared after treatment with saline (control), MTD and MET at 6 weeks (magnification ×100). b Bar graph shows the percentage of vWF-positive cells in the three groups (**P < 0.01 and ***P < 0.001)
Down-regulation of VEGFR-2 expression by MET chemotherapy
To investigate the anti-angiogenic effect by MET chemotherapy, the tumor tissues from the above-described experiments were collected and subjected to RT-PCR and western blotting to detect changes in VEGFR-2 expression. As shown in Fig. 6a, mRNA expression of VEGFR-2 was dramatically reduced by the MET chemotherapy. However, the level of VEGFR-2 mRNA expression after MTD treatment was not changed and comparable to that of the control group. In keeping with the results obtained with RT-PCR analysis, protein expression of VEGFR-2 was significantly down-regulated by MET chemotherapy compared with the control group, whereas the level of expression in the MTD group was similar to that seen with the control group (Fig. 6b). Figure 6c and d showed the band intensities on VEGFR-2 mRNA and protein expression, respectively (**P < 0.01 and ***P < 0.001).

Change in VEGFR-2 mRNA and protein expression with MET chemotherapy. a mRNA levels of expression of VEGFR-2 after treatment for 6 weeks. The expression of each lane was normalized to the level of GAPDH. RT-PCR was done in triplicate. b Protein expression of VEGFR-2 after treatment for 6 weeks. β-actin was used as a loading control. c and d Bar graph of the band intensities on VEGFR-2 mRNA and protein expression. VEGFR-2 mRNA and protein expression were quantified by densitometry with an Image software Gauge 4.0 program (**P < 0.01 and ***P < 0.001)
Discussion
Regardless of the etiology, the establishment of HCC is a result of a multistep carcinogenesis process that involves long-lasting inflammation in hepatocytes leading to cirrhosis. Commonly, this malignant tumor develops from a background of liver cirrhosis. In this study, we employed a DEN-induced cancer model which can establish HCC and liver cirrhosis simultaneously. Given that HCC occurs predominantly in cirrhotic livers, this HCC model may provide the similarity between experimental and human HCCs, being a better scheme for studying human HCC than the implanted HCC model without cirrhosis.
This study demonstrated that MET chemotherapy significantly inhibits growth and angiogenesis of tumors in a rat model of HCC. Furthermore, this type of treatment scheduling offered prolonged survival without major toxicities, as compared with MTD chemotherapy or untreated control groups. These findings add support for the potential therapeutic value of MET dosing in cancer therapy for patients with HCC.
CTX, an alkylating agent that is extensively used in the treatment of various malignant diseases, induces the formation of cross-links, which leads to replication arrest and cell death in cancer cells [22, 23]. Although both MET and MTD delivery of CTX have anti-cancer effects, earlier investigations have shown that tumor growth and angiogenesis were inhibited more with the MET delivery of CTX, which resulted in better antitumor efficacy and lower toxicity than MTD in various cancers other than HCC [24, 25]. In agreement with these observations, our data further confirmed the beneficial effect of MET dosing using CTX in a HCC model.
The primary mechanism for MET chemotherapy is considered to be inhibition of tumor angiogenesis by targeting endothelial cells in the tumor neovasculature [26]. Consistently, the data emerging from our DEN model showed that MET therapy using CTX induced a significant decrease in endothelial cell expression as compared with the MTD or control groups. Among VEGF receptors, VEGFR-2 plays a more functionally important role than VEGFR-1, because it can mediate signaling events including endothelial cell mitogenesis, migration, survival, and vascular invasion [27–29]. Our MET dosing in the DEN-induced HCC model significantly inhibited both mRNA and protein expression of VEGFR-2 after 6 weeks of therapy, whereas no changes in VEGFR-2 levels were observed with MTD therapy, again supporting the concept that this therapy works principally by blocking angiogenesis.
In the initial period of our experiments, MTD therapy appeared to work equally or slightly better in suppressing proliferation of tumor cells and increasing the apoptotic index than the MET CTX. Nevertheless, there was no beneficial effect of MTD therapy on anti-angiogenesis, which was clearly seen with the MET therapy during the same period. Killing only rapidly dividing cells by MTD therapy may be insufficient to treat all of tumor cells and the microenvironment, and consequently, there is potential for the development of resistance to chemotherapeutic agents [30, 31]. In this respect, it seems that the MET therapy is superior to the MTD regimen for inhibiting tumors, because it not only has a cytotoxic effect on tumor cells but also inhibits tumor endothelial cells. Although it was not evaluated in this study, the antitumor effect of MET therapy will become more apparent with more prolonged MET therapy beyond 6 weeks, without acquisition of drug resistance.
One can argue that the dose of CTX for MTD chemotherapy that we used was lower than the dose used in other studies [6, 17, 32], and this could be in part associated with the overall minor response for MTD therapy in our study. However, the dose (20 mg/kg twice a week) for MET therapy in this study was also lower compared with the MET dose in other experiments [3, 33]. Most of all, the total cumulative dose of CTX delivered was the same between the MTD and MET therapies, allowing more reliable interpretation of data results.
It has to be noted that the MTD schedule with a 21-day cycle of 40 mg/kg CTX 3 times a week was toxic, so that most rats in the MTD group experienced severe weight loss and a considerable proportion of them ultimately died before completion of the planned treatment. In fact, in our preliminary test using a higher dose of MTD, all of the rats died in the initial period of MTD therapy (data not shown). This seems to be a distinctive feature of the DEN-treated model in which liver cancer develops with associated liver cirrhosis. Even a modest chemotherapy schedule can cause a more severe local and systemic insult, leading to fatal outcome in this model. In contrast, the vast majority of subjects in the MET group tolerated the treatment schedule without significant weight loss or other signs of toxicity. Taken together, the reduced toxicity profiles and greater anti-angiogenic and antitumor efficacy of MET therapy would have contributed to prolong the survival in the present study.
In summary, the present study demonstrated that MET chemotherapy scheduling is not only at least as efficient as MTD therapy in suppressing the growth of tumor but also less toxic in a HCC model with associated liver cirrhosis. Furthermore, this type of therapy can effectively inhibit tumor endothelial cells. The overall findings suggest that MET chemotherapy has a high therapeutic value and should be considered for future clinical trials for patients with HCC.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- MTD:
-
Maximum tolerated dose
- MET:
-
Metronomic
- VEGF:
-
Vascular endothelial growth factor
- DEN:
-
Diethylnitrosamine
- CTX:
-
Cyclophosphamide
- PCNA:
-
Proliferating cell nuclear antigen
- vWF:
-
von-Willebrand factor
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- PAGE:
-
Polyacylamide gel electrophoresis
References
Block TM, Mehta AS, Fimmel CJ, Jordan R (2003) Molecular viral oncology of hepatocellular carcinoma. Oncogene 22:5093–5107
Song TJ, Ip EW, Fong Y (2004) Hepatocellular carcinoma: current surgical management. Gastroenterology 127:248–260
Bocci G, Francia G, Man S, Lawler J, Kerbel RS (2003) Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci USA 100:12917–12922
Hanahan D, Bergers G, Bergsland E (2000) Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest 105:1045–1047
Kerbel RS, Kamen BA (2004) The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 4:423–436
Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, Folkman J (2000) Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 60:1878–1886
Gasparini G (2001) Metronomic scheduling: the future of chemotherapy? Lancet Oncol 2:733–740
Stempak D, Seely D, Baruchel S (2006) Metronomic dosing of chemotherapy: applications in pediatric oncology. Cancer Invest 24:432–443
Giavazzi R, Taraboletti G (1999) Angiogenesis and angiogenesis inhibitors in cancer. Forum (Genova) 9:261–272
Risau W (1997) Mechanisms of angiogenesis. Nature 386:671–674
Veikkola T, Alitalo K (1999) VEGFs, receptors and angiogenesis. Semin Cancer Biol 9:211–220
Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J (2000) Vascular-specific growth factors and blood vessel formation. Nature 407:242–248
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3:401–410
Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6:389–395
Ferrara N, Gerber HP, Le Couter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676
Bertolini F, Paul S, Mancuso P, Monestiroli S, Gobbi A, Shaked Y, Kerbel RS (2003) Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res 63:4342–4346
Man S, Bocci G, Francia G, Green SK, Jothy S, Hanahan D, Bohlen P, Hicklin DJ, Bergers G, Kerbel RS (2002) Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res 62:2731–2735
Stolting S, Klink T, Bela C, Engels C, Wagner T (2004) Metronomic scheduling of trofosfamide chemotherapy in human NSCLC xenografts highly increases therapeutic efficacy compared to conventional scheduling by inhibition of angiogenesis. Int J Clin Pharmacol Ther 42:652–653
Lee TY, Kim KT, Han SY (2007) Expression of ErbB receptor proteins and TGF-alpha during diethylnitrosamine-induced hepatocarcinogenesis in the rat liver. Korean J Hepatol 13:70–80
Schiffer E, Housset C, Cacheux W, Wendum D, Desbois-Mouthon C, Rey C, Clergue F, Poupon R, Barbu V, Rosmorduc O (2005) Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 41:307–314
Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119:493–501
Pang LP, Huang W, Sun Q, Guo W, Li RT, Cui JR (2008) SLXM-2, a derivative of cyclophosphamide: mechanism of growth inhibition on hepatocarcinoma 22 cells. Anticancer Drugs 19:167–174
Lindner G, Botchkarev VA, Botchkareva NV, Ling G, van der Veen C, Paus R (1997) Analysis of apoptosis during hair follicle regression (catagen). Am J Pathol 151:1601–1617
Mirkes PE, Little SA (2000) Cytochrome c release from mitochondria of early postimplantation murine embryos exposed to 4-hydroperoxycyclophosphamide, heat shock, and staurosporine. Toxicol Appl Pharmacol 162:197–206
Shaked Y, Emmenegger U, Francia G, Chen L, Lee CR, Man S, Paraghamian A, Ben-David Y, Kerbel RS (2005) Low-dose metronomic combined with intermittent bolus-dose cyclophosphamide is an effective long-term chemotherapy treatment strategy. Cancer Res 65:7045–7051
Miller KD, Sweeney CJ, Sledge GW Jr (2001) Redefining the target: chemotherapeutics as antiangiogenics. J Clin Oncol 19:1195–1206
Meyer M, Clauss M, Lepple-Wienhues A, Waltenberger J, Augustin HG, Ziche M, Lanz C, Buttner M, Rziha HJ, Dehio C (1999) A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J 18:363–374
Rahimi N, Dayanir V, Lashkari K (2000) Receptor chimeras indicate that the vascular endothelial growth factor receptor-1 (VEGFR-1) modulates mitogenic activity of VEGFR-2 in endothelial cells. J Biol Chem 275:16986–16992
Veikkola T, Alitalo K (1999) VEGFs, receptors and angiogenesis. Semin Cancer Biol 9:211–220
Longley DB, Johnston PG (2005) Molecular mechanisms of drug resistance. J Pathol 205:275–292
Raguz S, Yague E (2008) Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer 99:387–391
Jia LJ, Wei DP, Sun QM, Jin GH, Li SF, Huang Y, Hua ZC (2007) Tumor-targeting Salmonella typhimurium improves cyclophosphamide chemotherapy at maximum tolerated dose and low-dose metronomic regimens in a murine melanoma model. Int J Cancer 121:666–674
Hermans IF, Chong TW, Palmowski MJ, Harris AL, Cerundolo V (2003) Synergistic effect of metronomic dosing of cyclophosphamide combined with specific antitumor immunotherapy in a murine melanoma model. Cancer Res 63:8408–8413
Acknowledgments
This research was supported by The Korean Association for the Study of the Liver, grant no. 0620390 from the National R&D Program for Cancer Control, Ministry for Health, Welfare and Family Affairs, Republic of Korea, and the second stage of Brain Korea 21 project.
Conflict of interest statement
There are no conflicts of interest in this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. T. Park and J. W. Jang equally contributed to this work.
Rights and permissions
About this article
Cite this article
Park, S.T., Jang, J.W., Kim, G.D. et al. Beneficial effect of metronomic chemotherapy on tumor suppression and survival in a rat model of hepatocellular carcinoma with liver cirrhosis. Cancer Chemother Pharmacol 65, 1029–1037 (2010). https://doi.org/10.1007/s00280-009-1108-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-1108-4