
Abstract
Purpose
The histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances cisplatin [cis-diammine dichloroplatinum (II)] (CDDP)-induced apoptosis in the oral squamous cell carcinoma (OSCC) cell line by complex, multifunctional mechanisms. We investigated the role of endoplasmic reticulum (ER) stress in the enhancing effect of SAHA on CDDP, compared with the ER stressor thapsigargin.
Methods
We chose OSCC cell line HSC-3 to ascertain the mechanism of SAHA-enhanced cytotoxicity among various cell lines. HSC-3 cells were incubated with CDDP/SAHA for 48 h, followed by the assessment of cell chemosensitivity to CDDP with MTT and TUNEL assays. Western blot analysis was used to detect the expressions of ER-related molecules, and flow cytometry was used to monitor caspase activity.
Results
Treatment with CDDP/SAHA potently induced apoptosis in HSC-3 cells with a significant increase in caspase-4 and -12 functions. For example, 60% of cells became apoptotic after 48 h of treatment with CDDP/SAHA. In addition, SAHA alone rapidly induced sustained phosphorylation of eukaryotic translation initiation factor-2 (eIF2)α, which is up-regulated during ER stress. Inhibition of ER stress by salubrinal, an inhibitor of eIF2α dephosphorylation, abrogated SAHA’s enhancement of CDDP cytotoxicity. Levels of phospho-Akt are decreased in SAHA-treated cells, and this is in turn associated with increased activity of protein phosphatase 1 (PP1) by SAHA, the phosphatase upstream of Akt.
Conclusion
These data indicate that up-regulation of specific-ER stress-associated events is an integral part of the mechanism by which SAHA enhances CDDP-induced apoptosis, and PP1 up-regulation followed by Akt dephosphorylation plays an important role in SAHA-enhanced CDDP apoptosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite the recently reported decrease in the overall death rate from cancer, the estimated survival rate and number of deaths from head and neck cancer, including the oral cavity, oropharynx, larynx, hypopharynx, and nasopharynx, remain virtually unchanged [1]. Chemotherapy is especially active in oral cavity cancers and so the development of a chemotherapeutic strategy with tumor specificity to decrease drug side effects and/or to increase anti-tumor efficacy is urgently required. Cisplatin [cis-diammine dichloroplatinum (II)] (CDDP) and its analogues are particularly useful in the treatment of late-stage oral squamous cell carcinoma (OSCC), and are included in the standard chemotherapy treatment for this disease [2]. It is believed that DNA platination and the subsequent induction of apoptosis is the primary cytotoxic mechanism of CDDP, although the mechanism whereby these DNA adducts kill cells is not fully understood. Unfortunately, significant levels of resistance in OSCC cells occur rapidly following CDDP treatment, resulting in failure to increase survival when administered as monotherapy or in combination with surgery or radiotherapy [3].
In the last few years, our research has aimed to establish a theoretical base for the enhancement of CDDP-induced apoptosis by epigenetic agents in human OSCC cells. We have reported that the histone deacetylase (HDAC) inhibitor, suberoylanilide hydroxamic acid (SAHA, generic name Vorinostat), was capable of enhancing apoptosis in CDDP-treated OSCC cells [4]. SAHA was the first HDAC inhibitor to be approved by the US Food and Drug Administration for cutaneous T-cell lymphoma in 2006. These results demonstrated that intracellular-reduced glutathione plays a key role in governing SAHA-dependent enhancement of CDDP-induced apoptosis; however, it seems likely that other crucial mechanisms may be required to mediate the enhancement of CDDP-induced apoptosis by SAHA. An additional mechanism by which CDDP can cause apoptosis is known to be through the endoplasmic reticulum (ER) stress pathway [5]. There is currently strong interest in assessing whether the mechanism of SAHA enhancement involves potentiation of the ER stress response.
Apoptotic cell death can be initiated by extrinsic (receptor-mediated) or intrinsic (organelle-mediated) signaling pathways. Recently, it has been shown that disturbances in ER homeostasis lead to an evolutionarily conserved cell response (ER stress) [6]. Prolonged ER stress impairs the protective mechanisms designed to promote correct folding and to degrade faulty proteins, ultimately leading to organelle dysfunction and apoptotic cell death [7]. ER homeostasis can be disrupted by numerous factors, including the accumulation of unfolded and misfolded proteins, calcium depletion, glucose deprivation, accumulation of free cholesterol, and viral infection [8, 9]. Recent reports suggested that HDAC inhibitors have been reported to induce the accumulation of misfolded proteins [10, 11]; therefore, HDAC inhibitors represent yet another class of chemotherapeutic drugs, which can trigger ER stress-induced apoptosis of tumor cells by causing the accumulation of misfolded proteins.
Many cellular components with nucleophilic sites, such as DNA, RNA, proteins, membrane phospholipids, cytoskeletal microfilaments, and thiol-containing molecules, react with CDDP, where only approximately 1% of the intracellular CDDP reacts with nuclear DNA to yield a variety of adducts that include interstrand and intrastrand DNA cross-links and DNA-protein cross-links [12]. There is evidence that other cellular targets may also be involved in drug cytotoxicity. Recent studies showed that CDDP cytotixicity is involved in the ER stress response in both cancer cells [5] and normal kidney proximal tubule cells [13]. CDDP increased caspase-12 cleavage (ER stress-specific) in head and neck cancer cells [14]. In CDDP-induced apoptosis of melanoma cells, calpain, independent of Ca2+ imbalance, is activated upstream of effector caspases and plays a major role in cell death [15]. Activation of the unfolded protein response (UPR) is involved in the resistance of melanoma cells to CDDP, and this is associated with 78-kDa glucose-regulated protein (GRP78)-mediated inhibition of the activation of caspase-4 and -7. In either case, these results provide new insights into the apoptotic mechanisms of CDDP.
Although DNA lesions have been presumed to be the predominant mechanism for CDDP-induced cytotoxicity, apoptosis can also be initiated through the ER stress pathway. Here, we investigate damage to the ER after CDDP treatment combined with SAHA, compared with each alone. Our results revealed that CDDP/SAHA induced atypical ER stress responses in OSCC cells, providing novel insights into the SAHA-mediated enhancement of anti-tumor activity by CDDP.
Materials and methods
Reagents
Cisplatin [cis-diammine dichloroplatinum (II)] (Nippon Kayaku, Tokyo, Japan) was used in this study. SAHA was obtained from ALEXIS (Lausen, Switzerland). Etoposide (VP16) and valproic acid (VPA) were purchased from Sigma (St. Louis, MO). ER stressors, thapsigargin (TG), and tunicamycin (TM) were purchased from Calbiochem (La Jolla, CA). ER stress inhibitor, salubrinal (Sal), calpain inhibitor SJA6017 (SJA), c-Jun NH2 terminal kinase (JNK) inhibitor SP600125 (SP), nuclear inhibitor of protein phosphatase-1 (NIPP-1), and phosphoinositide 3-kinase (PI3K) inhibitor LY294002 (LY) were purchased from Calbiochem. Inhibitors of the mitochondrial permeability transition pore, cyclosporin A (CsA) and bongkrekic acid (BA) were obtained from Biomol (Plymouth Meeting, PA) and Calbiochem, respectively. All other chemicals used in this study were commercially available.
Caspase inhibitors
Inhibitors of pan-caspase (z-VAD-fmk), caspase-3 (z-DEVD-fmk), caspase-4 (z-LEVD-fmk), and caspase-12 (z-ATAD-fmk) were purchased from MBL (Nagoya, Japan). Cells were pretreated with 20 μM of these inhibitors for 1 h at 37°C before each treatment. A DMSO control was also included as a control for the given concentration of each inhibitor.
Cell lines and cell culture
HSC-3 cells, HSC-4 cells (mutant-type p53, human oral squamous cell carcinoma), A549 cells (wild-type p53, human non-small-cell lung adenocarcinoma), and MCF-7 cells (wild-type p53, human breast cancer) were obtained from the Cell Resource Center for Biomedical Research (Institute of Development, Aging and Cancer, Tohoku University, Japan). Cells were cultured in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). Cells were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Cell viability assays
Cell viability was evaluated by the trypan blue exclusion assay and then the cytotoxic effects of drugs were determined using an MTT (WST-8) colorimetric assay kit (Dojindo, Kumamoto, Japan) as previously described [4]. IC20 values that cause 20% cell-growth inhibition were determined after 48-h treatments.
Measurement of intracellular caspase activity
Intracellular caspase-4 activity was measured using the labeled substrate LEVD-p-nitroanilide (pNA) (MBL). Extracts from cells stimulated with the indicated agents were assayed, and absorbance was measured at 405 nm in a microplate reader. Intracellular caspase-12 activity was measured using FITC-ATAD-fmk (Abcam, Cambridge, MA). The inhibitor binds irreversibly to activated caspases, enabling measurement of active caspase within the cell. Labeled cells were analyzed by flow cytometry to determine the percentage of intracellular active cells for caspase-12.
TUNEL assay
Apoptotic cells were assayed by the TUNEL method using the Mebstain apoptosis kit direct (MBL) for flow cytometric analysis (FACSCalibur; Becton-Dickinson, San Jose, CA).
Protein phosphatase activity
Protein phosphatase 1 (PP1) activity was analyzed using p-nitrophenyl phosphate (pNPP) as a colorimetric phosphatase substrate (AnaSpec, San Jose, CA). According to the manufacturer’s instructions, whole cell protein was extracted, and equal amounts of proteins were incubated with pNPP reaction mixture for 1 h at room temperature. Stop solution was added, and absorbance was measured at 405 nm in a microplate reader.
Western blotting
Whole or nuclear proteins were analyzed by Western blotting. Rabbit polyclonal anti-GRP78 antibody was purchased from AnaSpec. Rabbit polyclonal anti-GRP94, anti-phospho-eIF2α (Ser51), and anti-total-eIF2α antibodies were purchased from Cell Signaling (Danvers, MA). Goat polyclonal anti-GADD34 antibody was obtained from Abcam. Mouse monoclonal anti-β-actin antibody was purchased from BioVision (Mountain View, CA). HRP-conjugated secondary antibodies, sheep anti-mouse and donkey anti-rabbit antibody (GE Healthcare, Piscataway, NJ), and mouse anti-goat antibody (Assay designs, Ann Arbor, MI) were used.
Statistical analysis
Data are given as the mean ± SE. Multiple comparisons were made by Scheffe’s test. P values <0.05 were regarded as significant.
Results
Enhancement of CDDP-induced apoptosis by SAHA
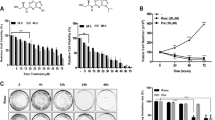
To further characterize SAHA-mediated apoptotic enhancement [4], several cell lines were treated for 48 h with CDDP in the presence of SAHA, and analyzed by the TUNEL method that measures DNA fragmentation. When two cell lines (HSC-3 and MCF-7) were treated with CDDP/SAHA, the level of apoptosis showed significant enhancement among the cell lines tested (Fig. 1a). In addition, HSC-3 cells were treated with CDDP in the presence of another HDAC inhibitor, VPA, and evaluated for apoptotic cells. The average percentage of apoptotic cells was higher in cultures treated with CDDP/SAHA together, compared with CDDP/VPA (Fig. 1b).

Enhancement of CDDP-induced apoptosis by SAHA. a Cancer cell lines (5 × 105 cells/well) were treated with 5 μg/ml CDDP for 48 h with or without SAHA (IC20: 1.5–6.0 μM), followed by TUNEL assay. Results are expressed as the mean intensity of fluorescence (MIF) of triplicate experiments. b HSC-3 cells were treated with CDDP for 48 h in the presence or absence of SAHA (1.5 μM) or VPA (1 mM), followed by TUNEL assay. Percentages of apoptosis cells treated with each alone were 5.9 ± 0.7% (SAHA) and <0.1% (VPA). c HSC-3 cells were treated with CDDP for 24 h in the presence or absence of TG (0.2 μM) or TM (2 μg/ml), followed by TUNEL assay. Percentages of apoptosis in each alone were 0.8 ± 0.3% (TG) and 1.7 ± 0.9% (TM). Results represent the mean ± SE of triplicate experiments. *P < 0.01 compared to CDDP alone
To compare with ER stress-induced apoptosis, HSC-3 cells were treated with CDDP in combination with the representative ER stressors, TG, and TM. ER stress can be experimentally induced by treating cells with TG, which depletes intracellular calcium stores or TM, which inhibits protein glycosylation. Although the stressor alone did not induce apoptosis significantly, treatment with stressor TG was shown to enhance CDDP-induced apoptosis as well as CDDP/SAHA treatment (Fig. 1c).
Role of ER-associated caspases in CDDP/SAHA-induced apoptosis
To confirm the involvement of ER stress in the response of HSC-3 cells to CDDP/SAHA, we analyzed caspase-4 and -12 activities, which are causally involved in triggering apoptosis under ER stress. Human ER-specific caspase-4 was activated after CDDP/SAHA treatment, as expected (Fig. 2a). FITC-labeled small-molecule inhibitor of the active caspase-12 was used to measure caspase-12 cleavage after treatment with CDDP/SAHA. Using flow cytometry, we observed that CDDP/SAHA caused ER stress-induced apoptosis (caspase-12 cleavages) that contributed to cellular apoptosis (Fig. 2b). Notably, after treatment with SAHA alone there were significant increases in caspase-12 activity (Fig. 2c), indicating its cleavage into active caspase-12, usually induced by ER stress.

Role of ER-associated caspases in CDDP/SAHA-induced apoptosis. a–c HSC-3 cells were treated with CDDP or SAHA (5 μM or indicated doses) for 24 h. Intracellular caspase activities were measured using LEVD-pNA for caspase-4 (a, c) and FITC-ATAD-fmk for caspase-12 (b, c). Caspase-4 activity is expressed as OD405 (a) or relative values (c). Caspase-12 activity is expressed as MIF (b) or relative MIF (c). d Cells pretreated for 1 h with each caspase inhibitor (20 μM) or DMSO alone (vehicle) were treated for 24 h with CDDP/SAHA or CDDP/TG, and evaluated by TUNEL assay. CDDP/SAHA or CDDP/TG treatments are expressed as 100, respectively, and the effect of each caspase inhibitor is shown as a relative ratio. Results are expressed as the mean ± SE of triplicate experiments. P < 0.05 compared to CDDP/SAHA (#) or CDDP/TG (*)
Next, we used specific caspase inhibitors to confirm the caspases involved in the induction of apoptosis by CDDP/SAHA. HSC-3 cells were pretreated for 1 h with a pan-caspase inhibitor or inhibitors of caspase-3, -4, -8, -9, or -12, followed by the treatment with CDDP/SAHA or CDDP/TG (Fig. 2d) for 24 h before TUNEL staining. The broad-spectrum caspase inhibitor z-VAD-fmk almost completely inhibited apoptosis induced by CDDP/SAHA (Fig. 2d). Caspase-12 (z-ATAD-fmk) and -4 (z-LEVD-fmk) inhibitors substantially inhibited CDDP/SAHA-induced apoptosis, whereas higher inhibitory effects against CDDP/TG-induced apoptosis were observed in each inhibitor. By contrast, the addition of inhibitors of caspase-8 (z-IETD-fmk) or caspase-9 (z-LEHD-fmk) had only small effects on the induction of apoptosis by CDDP/SAHA (data not shown). The caspase-3 inhibitor slightly inhibited apoptotic cell death at the 24-h time point in response to CDDP/SAHA or CDDP/TG. These data suggest that synergistic death induced by CDDP/SAHA is dependent on ER-associated caspase activation as well as CDDP/TG.
Inhibition of CDDP/SAHA-induced apoptosis by salubrinal
To verify the significance of the role played by mitochondria in CDDP/SAHA-induced apoptosis, HSC-3 cells were pretreated with the classical blockers of mitochondrial permeability transition pores, CsA and BA, resulting in the inhibition of both mitochondria depolarization and cytochrome c release. The results in Fig. 3a and b indicate that the mitochondrial pathway accounts only partially for CDDP/SAHA- and CDDP/TG-induced apoptosis, suggesting that additional mechanisms are also involved. Both the μ-calpain inhibitor SJA6017, and the JNK inhibitor SP600125 significantly reduced apoptotic rates in cells treated with CDDP/TG, but neither inhibitor induced a reduction in cells treated with CDDP/SAHA. Taken together, these results imply that the induction of apoptosis with CDDP/SAHA is caused by calpain-independent caspase-12 activation.

Inhibition of CDDP/SAHA-induced apoptosis by salubrinal. a, b HSC-3 cells pretreated for 1 h with salubrinal (Sal, 50 μM), NIPP-1 (2.5 pM), SJA6017 (SJA, 50 μM), SP600125 (SP, 50 μM), cyclosporine A (CsA, 0.25 μM) or bongkrekic acid (BA, 0.25 μM) were treated for 24 h with CDDP combined with 1.5 μM SAHA (a) or 0.2 μM TG (b), followed by TUNEL assay. Results represent the mean ± SE of triplicate experiments. *P < 0.05 compared to CDDP/SAHA or CDDP/TG. c, d Cells were pretreated for 1 h with Sal, followed by treatment of VP16 (10 μM) combined with SAHA (c) or TG (d) for 24 h. Percentages of apoptosis cells were determined
We examined the effect of PP1 inhibitors on CDDP/SAHA-induced apoptosis. The specific eIF2α phosphatase inhibitor, Sal, and a nuclear inhibitor of PP1, NIPP1, were used. Salubrinal, but not NIPP1 treatment significantly abolished the apoptosis observed after treatment with CDDP/SAHA (Fig. 3a) or CDDP/TG (Fig. 3b). Inhibition of CDDP/TG-induced apoptosis by Sal exceeded that by z-VAD-fmk, indicating that the enhanced apoptosis by CDDP/SAHA was due primarily to ER stress-induced apoptosis because Sal is specific to the inhibition of ER stress-induced cell death; however, CDDP/SAHA-induced apoptosis was attributable to atypical ER stress distinct from the classic ER stress inducer TG. We also assessed the effect of Sal on other DNA-damaging agent. HSC-3 cells treated for 24 h with etoposide (10 μM)/SAHA (Fig. 3c) or etoposide/TG (Fig. 3d) in the presence of Sal showed no inhibition of apoptotic efficiency.
Expression of ER-associated molecules in CDDP or SAHA stimulation
Exposure to CDDP or SAHA for various time periods revealed that protein levels of ER stress markers, GRP78 and GRP94, were significantly increased in TG- and TM-treated cells, but not in CDDP- or SAHA-treated cells (Fig. 4a). Furthermore, we tested the expression and phosphorylation levels of eIF2 known to be involved in translation initiation. As shown in Fig. 4b, exposure to SAHA alone resulted in the rapid phosphorylation of eIF2α, an event that was sustained over the ensuing 24 h. In contrast, no major changes were detected in eIF2α total protein levels.

Expression of ER-associated molecules in CDDP or SAHA stimulation. a, b HSC-3 cells were treated with CDDP (5 μg/ml), SAHA (5 μM), TG (2 μM) or TM (2 μg/ml) for 48 h for GRP78 and GRP94 (a), and for 6 h for eIF2α (b). Total or nuclear cell extracts were prepared, and equal amounts of extracts were loaded for Western blotting analysis. β-actin was used as a loading control
Activation of PP1 by SAHA treatment
We measured PP1 activity in HSC-3 cell lysates treated with SAHA and/or Sal. As shown in Fig. 5a, SAHA treatment increased phosphatase activity in a time-dependent manner, and the addition of Sal decreased phosphatase activity at 3-h incubation. Taken together, these results clearly demonstrated that SAHA enhancement of the CDDP apoptotic effect on OSCC cells was in part due to the activation of PP1 activity induced by SAHA treatment. To show that there was a relationship between the up-regulation of PP1 and down-regulation of Akt phosphorylation, HSC-3 cells were treated for 4 h with SAHA alone. Dephosphorylation of Akt was observed by treatment with SAHA or PI3K inhibitor LY294002 (Fig. 5b). There was no decrease in total Akt after either treatment. Finally, Akt phosphorylation was down-regulated by SAHA, which was accompanied with an increase in phospho-eIF2α by SAHA (Fig. 4b).

Activation of PP1 and dephosphorylation of Akt by SAHA. a HSC-3 cells were treated with SAHA (5 μM) with or without Sal (50 μM). Whole cell proteins were extracted, and equal amounts of extracts were incubated with pNPP reaction mixture for 1 h. Results represent OD405 values. b Cells were cultured for 4 h with or without SAHA (1.5 and 5.0 μM) or LY294002 (LY, 30 μM), and fixed to measure total and phosphorylated Akt. Results represent the mean ± SE of triplicate values of OD450. *P < 0.05 compared to the untreated control
Discussion
In this work, we investigated the involvement of ER stress in the initiation of apoptosis elicited by CDDP/SAHA combination, because cell death is conspicuous in cells that have encountered insurmountable ER stress. First, we compared the effect of CDDP/SAHA on four cancer cell lines originating from different organs containing wild-type or mutant-type p53. CDDP/SAHA together induced a higher level of apoptosis in HSC-3 and MCF-7 cells compared with other cell lines tested (HSC-4 or A549). There was no parallel between SAHA and p53 status in CDDP-induced apoptosis. We observed a significant increase in the phosphorylation of eIF2α and caspase-4 and -12 functions in HSC-3 cells that demonstrated enhancement of CDDP-induced cytotoxicity by SAHA. Our data demonstrating that VPA is much less potent as an HDAC inhibitor than SAHA (mM vs. μM, respectively) was, in part, attributable to the inability to access the zinc cation in the HDAC active site pocket, which is pivotal to deacetylation catalysis [16, 17]. In addition, the effects of SAHA differ from those elicited by classical ER stress inducers (TM and TG) in that SAHA are not associated with the activation of calpain and JNK.
It is noteworthy that for drugs such as CDDP and some other alkylating agents, UPR induction leads to enhanced drug sensitivity. By contrast, the DNA-damaging topoisomerase II inhibitor etoposide, which also disrupts mitochondrial activity, does not induce apoptotic signaling in ER stress in cells. The ER takes part in initiating apoptosis through different mechanisms, which may activate caspase-12 [7, 18]. Intracellular calcium increment causes the activation of μ-calpain followed by caspase-12 activation. Apoptosis induced by CDDP/SAHA has been shown to involve the activation of caspase-12, although the human caspase-12 gene contains severe mutations which preclude its function as a regular caspase [19]. We attempted combined treatment of CDDP/SAHA-treated cells with BAPTA-AM (a chelator of intracellular Ca2+); however, the combined treatment by itself had a cytotoxic effect on HSC-3 cells. Unlike CDDP/TG stimulation, μ-calpain inhibitor was not effective in CDDP/SAHA stimulation, suggesting that ER stress markers and ER-mediated cell death signaling pathways are not always altered to the same extent. This result further suggests that, in our model, mechanisms other than Ca2+-imbalance and calpain activation are mainly involved in CDDP/SAHA-induced apoptosis, which could cause apoptosis directly or through unknown intermediates.
Recently, a human colon cDNA library was screened using the sequence of mouse caspase-12 as a probe, and it was found that caspase-4 is a human candidate of ER-dependent caspase [20]. Caspase-4 on the ER is supposed to function in ER stress-induced apoptosis similarly to caspase-12. Treatment with caspase-4 inhibitor significantly increased resistance to CDDP/SAHA- or CDDP/TG-induced cell death. It is possible that both caspases-12 and -4 are involved in our experimental model using human OSCC cells. Because the A549 cell line is capable of undergoing ER stress-induced apoptosis through caspase-12 cleavage when treated with other agents [21], the low apoptotic efficiency observed following treatment with CDDP is not due to an intrinsic defect in this pathway in this cell line. Cyclosporin A and BA (mitochondrial permeability transition pore inhibitors) did not display significant inhibition of CDDP/SAHA-induced apoptotic cell death. Further studies, however, are required to clarify the involvement of mitochondria during the CDDP/SAHA-induced apoptosis of HSC-3 cells.
ER stress involves the phosphorylation of eIF2α on Ser51 by ER-localized kinases [22]. The inactivation of RNA-dependent protein kinase-like ER kinase (PERK) or the inhibition of eIF2α phosphorylation has been found to promote ER stress-mediated apoptosis [23, 24]. In contrast, ER stress can also potently induce apoptosis by inducing pro-apoptotic genes in response to ER stress [25, 26]. Salubrinal inhibits the PP1-mediated dephosphorylation of eIF2α but not the dephosphorylation of many other PP1 substrates in the cell [27]. Indeed, we observed that Sal inhibits the activity of serine/threonine phosphatase PP1 induced by SAHA, and protect cells against apoptotic stimuli (CDDP/SAHA) related to ER stress. SAHA induced eIF2α phosphorylation, and enhanced the apoptotic potential of CDDP. HDAC inhibitors (SAHA and TSA) facilitate the dephosphorylation of Akt by altering the dynamics of HDAC-PP1 complexes and increasing the association of PP1 with Akt [28]. It was also observed that TG treatment caused a decrease in phospho-Akt levels in human renal carcinoma Caki cells [29]. Akt activation, which is mediated through phosphorylation, maintains a survival signal that protects cells from apoptosis by phosphorylating pro-apoptotic proteins, such as caspase-9, Bad, and the cell regulatory protein GSK3β [30]. A recent study also showed that inactivation of PI3K/Akt is important in ER stress-induced apoptosis, where TM induces cell death, suppressing Akt activity by lowering its protein expression level [31]. It is becoming accepted that an important cause of CDDP resistance relates to aberrant functioning of the apoptotic mechanism in cancer cells with Akt-regulated survival pathways [32]; therefore, suppression of Akt phosphorylation may be an important mechanism associated with CDDP/SAHA-induced apoptotic cell death. This histone-independent mechanism provides a potential basis to account for the anti-neoplastic activities of these agents in growth inhibition and apoptosis induction. SP600125 blocked apoptosis induced by CDDP/TG, but not CDDP/SAHA, indicating that JNK activation plays an important role in initiating classical ER stress-mediated apoptosis [33].
Although CDDP remains a vital mainstay of chemotherapy, the incidences of recurrence and platinum resistance lead to poor survival rates for head and neck cancer patients, and underscore the need to develop effective modulators of platinating agents. In this study, we have shown that SAHA enhances the efficacy of CDDP by activating the atypical ER stress pathway to increase apoptosis, where SAHA acts as an ER-stress mediator and apoptosis enhancer. Further investigation will yield information about the targets of CDDP and SAHA in this pathway.
References
Licitra L, Grandi C, Guzzo M, Mariani L, Lo Vullo S, Valvo F, Quattrone P, Valagussa P, Bonadonna G, Molinari R, Cantu G (2003) Primary chemotherapy in resectable oral cavity squamous cell cancer: a randomized controlled trial. J Clin Oncol 21:327–333
Foote RL, Kasperbauer JL, Okuno SH, Nichols DA, Olsen KD, Brown PD, Garces YI, Sargent DJ, Strome SE (2005) A pilot study of high-dose intraarterial cisplatin chemotherapy with concomitant accelerated radiotherapy for patients with previously untreated T4 and selected patients with T3N0-N3M0 squamous cell carcinoma of the upper aerodigestive tract. Cancer 103:559–568
Akervall J, Guo X, Qian CN, Schoumans J, Leeser B, Kort E, Cole A, Resau J, Bradford C, Carey T, Wennerberg J, Anderson H, Tennvall J, Teh BT (2004) Genetic and expression profiles of squamous cell carcinoma of the head and neck correlate with cisplatin sensitivity and resistance in cell lines and patients. Clin Cancer Res 10:8204–8213
Rikiishi H, Shinohara F, Sato T, Sato Y, Suzuki M, Echigo S (2007) Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. Int J Oncol 30:1181–1188
Mandic A, Hansson J, Linder S, Shoshan MC (2003) Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem 278:9100–9106
Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63
Rao RV, Hermel E, Castro-Obregon S, Del Rio G, Ellerby LM, Ellerby HM, Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program mechanism of caspase activation. J Biol Chem 276:33869–33874
Maxfield FR, Tabas I (2005) Role of cholesterol and lipid organization in disease. Nature 438:612–621
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115:2656–2664
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115:727–738
Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K Jr, Pal A, Bornmann WG, Chiao PJ, Huang P, Xiong H, Abbruzzese JL, McConkey DJ (2006) Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res 66:3773–3781
Gonzalez VM, Fuertes MA, Alonso C, Perez JM (2001) Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol 59:657–663
Liu H, Baliga R (2005) Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol 16:1985–1992
Rabik CA, Fishel ML, Holleran JL, Kasza K, Kelley MR, Egorin MJ, Dolan ME (2008) Enhancement of cisplatin cytotoxicity by O6-benzylguanine involves endoplasmic reticulum stress. J Pharmacol Exp Ther 327:442–452
Del Bello B, Moretti D, Gamberucci A, Maellaro E (2007) Cross-talk between calpain and caspase-3/-7 in cisplatin-induced apoptosis of melanoma cells: a major role of calpain inhibition in cell death protection and p53 status. Oncogene 26:2717–2726
Marks PA, Miller T, Richon VM (2003) Histone deacetylases. Curr Opin Pharmcol 3:344–351
Lu Q, Yang YT, Chen CS, Davis M, Byrd JC, Etherton MR, Umar A, Chen CS (2004) Zn2+-chelating motif-tethered short-chain fatty acids as a novel class of histone deacetylase inhibitors. J Med Chem 47:467–474
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403:98–103
Fischer H, Koenig U, Eckhart L, Tschachler E (2002) Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun 293:722–726
Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M (2004) Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol 165:347–356
Bitko V, Barik S (2001) An endoplasmic reticulum-specific stress-activated caspase (caspase-12) is implicated in the apoptosis of A549 epithelial cells by respiratory syncytial virus. J Cell Biochem 80:441–454
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176
Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol 5:781–792
Yamaguchi H, Bhalla K, Wang H (2003) Bax plays a pivotal role in thapsigargin-induced apoptosis of human colon cancer HCT116 cells by controlling Smac/Diablo and Omi/HtrA2 release from mitochondria. Cancer Res 63:1483–1489
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J (2005) A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307:935–939
Chen CS, Weng SC, Tseng PH, Lin HP, Chen CS (2005) Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem 280:38879–38887
Lee TJ, Lee JT, Kim SH, Choi YH, Song KS, Park JW, Kwon TK (2008) Overexpression of Par-4 enhances thapsigargin-induced apoptosis via down-regulation of XIAP and inactivation of Akt in human renal cancer cells. J Cell Biochem 103:358–368
Querfeld C, Rizvi MA, Kuzel TM, Guitart J, Rademaker A, Sabharwal SS, Krett NL, Rosen ST (2006) The selective protein kinase Cβ inhibitor enzastaurin induces apoptosis in cutaneous T-cell lymphoma cell lines through the AKT pathway. J Invest Dermatol 126:1641–1647
Yung HW, Korolchuk S, Tolkovsky AM, Charnock-Jones DS, Burton GJ (2007) Endoplasmic reticulum stress exacerbates ischemia-reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J 21:872–884
Asselin E, Mills GB, Tsang BK (2001) XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res 61:1862–1868
Lei P, Abdelrahim M, Cho SD, Liu X, Safe S (2008) Structure-dependent activation of endoplasmic reticulum stress-mediated apoptosis in pancreatic cancer by 1, 1-bis(3′-indoly)-1-(p-substituted phenyl)methanes. Mol Cancer Ther 7:3363–3372
Acknowledgments
We thank Mr. D. Mrozek for editing the manuscript. This work was supported in part by Grant-in-Aids for Scientific Research (20659309) from the Japan Society for the Promotion of Science and (19791480 and 19791482) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Suzuki, M., Endo, M., Shinohara, F. et al. Enhancement of cisplatin cytotoxicity by SAHA involves endoplasmic reticulum stress-mediated apoptosis in oral squamous cell carcinoma cells. Cancer Chemother Pharmacol 64, 1115–1122 (2009). https://doi.org/10.1007/s00280-009-0969-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-0969-x