
Abstract
Purpose
This is a phase-I study of gefitinib in combination with temozolomide in patients with gliomas. The goal of the study was to define the maximum tolerated dose (MTD) and to characterize the pharmacokinetics of gefitinib when combined with temozolomide.
Patients and methods
Patients were stratified according to co-administration of enzyme-inducing anti-epileptic drugs (EIAEDs). There were 26 evaluable patients enrolled (16 on EIAEDs, 10 not on EIAEDs). All but seven patients had Glioblastoma Multiforme (GBM), and only six cases had a Karnosfsky Performance Status (KPS) of less than 80; median age was 51 years. All had received prior radiotherapy and 14 patients had no prior chemotherapy. The starting dose of temozolomide was 150 mg/m2/day for 5 days every 28 days and could be escalated to a maximum dose of 200 mg/m2/day in subsequent cycles. The starting dose of gefitinib was 500 mg/day given by mouth on a continuous basis. Dose-limiting toxicity was assessed in cycle one only.
Results
For patients on EIAEDs, the MTD of gefitinib was 1,000 mg/day in combination with temozolomide. Dose-limiting toxicity (DLT) was due to diarrhea, nausea and vomiting. For patients not on EIAEDs, the MTD was 250 mg/day in combination with temozolomide. The DLT was due to increases in liver transaminases. Rash was not a significant toxicity at these dose levels. The peak concentration and AUC0-24hr at the 500 mg dose level was 1.8 and 2.5-fold lower, respectively, in the EIAED group compared to the non-EIAED group; trough levels of gefitinib increased in both groups consistent with the reported terminal half-life ranging from 27 to 51 h.
Conclusion
The recommended phase-2 dose of gefitinib when used in combination with temozolomide is 1,000 and 250 mg/day, respectively, for patients on or not on EIAEDs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The epidermal growth factor receptor (EGFR) is amplified in 40–50% of patients with Glioblastoma Multiforme (GBM), the most lethal of the malignant gliomas [3]. Over-expression also occurs, in some cases independent of gene amplification. In addition, there are mutant forms of the receptor, including the EGFRvIII mutant [3]. Epidermal growth factor (EGF), the ligand for the receptor, has been shown to stimulate tumor growth and migration in animal model systems, effects which can be abrogated by anti-EGFR monoclonal antibodies. These pre-clinical results supported the use of EGFR targeted treatment strategies for these tumors, including the use of small molecule receptor tyrosine kinase inhibitors. Two kinase inhibitors have been used clinically in malignant glioma, gefitinib (Iressa, ZD1839; AstraZeneca) and erlotinib (Tarceva, OSI-774; Genentech). Only modest benefits have been reported when used as single agents in recurrent malignant glioma, with few objective responses, although some benefit in delay of disease progression have been noted [8, 17].
There may be an advantage to use EGFR-targeted therapies earlier in the disease, and in combination with cytotoxic agents. The current standard for treatment of newly diagnosed GBM is surgery followed by concurrent radiotherapy and temozolomide chemotherapy, followed by adjuvant temozolomide for up to 6 months [20]. Despite this approach, median survival remains only 14.6 months. Although of limited benefit in recurrent disease, inhibitors of EGFR may be more efficacious in newly diagnosed disease. It has been shown that EGFR expression may be reduced at the time of tumor progression when compared to newly diagnosed, untreated disease [18]. It has also been shown that radiotherapy may increase EGFR activation, and EGFR over-expression has been shown in pre-clinical models to increase resistance to that treatment [5, 19]. In addition, experiments using human tumor xenograft models of GBM implanted in nude mice suggest that EGFR amplification confers resistance to alkylator-based treatment, including temozolomide [9]. These pre-clinical data suggest that inhibition of the EGFR early in the disease process, when expression may be higher, may potentially improve the response to radiation and/or chemotherapy given concurrently with radiation and following radiation.
Gefitinib, like erlotinib, is metabolized by CYP3A4, and exposure is reduced with concurrent use of enzyme inducing drugs. Stratification of patients based upon use of enzyme inducing agents is important to understand the toxicity and exposure of both tyrosine kinase inhibitors in this patient population. A previous phase-1 trial of erlotinib plus temozolomide has recently been reported and a recommended phase-2 dose of that regimen has been suggested [15]. The present study evaluates the toxicity and pharmacokinetics of the combination of gefitinib with temozolomide in patients with stable or recurrent malignant glioma, with and without the use of enzyme inducing antiepileptic drugs (EIAEDs). The goal of this study was to establish a recommended phase-2 dose for this combination that may be used for future studies in primary, newly diagnosed GBM, or as a component of multimodality treatment in recurrent disease.
Patients and methods
Patients aged ≥18 years with histologically proven supratentorial malignant glioma, including GBM, anaplastic astrocytoma, anaplastic oligodendroglioma, anaplastic mixed oligoastrocytoma, and malignant astrocytoma not otherwise specified, were eligible for inclusion. A life expectancy of >8 weeks and a KPS of ≥60 were required.
Patients with either stable or progressive disease were eligible for treatment. Adequate bone marrow (white blood cells >3,000/μl, absolute neutrophil count >1,500/μl, platelet count >120,000/μl, hemoglobin >10 g%), liver (serum glutamic–oxaloacetic transaminase and bilirubin <1.5 times the upper limit of normal) and renal function (creatinine <1.5 mg/dl or estimated creatinine clearance > 60 ml/min) were required within 14 days before entering the study.
Patients must have recovered from toxic effects of prior therapy with a minimum of 2 weeks after receiving vincristine, 6 weeks for nitrosoureas, 4 weeks for temozolomide, 3 weeks for procarbazine and 1 week for non-cytotoxic agents. Radiation had to be completed at least 3 weeks before study entry. Patients had to be at least 7-day post-tumor resection, if done, and recovered from the effects of surgery or radiation. Measurable disease following recent resection of a recurrent tumor was not required for study entry. No more than three prior chemotherapeutic regimens were permitted. Patients also had to be on stable doses of concomitant medication, especially those affecting the cytochrome P450 pathway (a minimum of 2 weeks), before entering the study. Growth factors or other investigational drugs was not permitted nor was previous use of gefitinib or erlotinib or any other anti-EGFR directed therapies. Exclusion criteria included patients with major illness inadequately controlled with therapy or disease that would compromise their ability to tolerate study treatment; significant gastrointestinal risk factors within the past 6 months; other cancers (except non-melanoma skin cancer or carcinoma in situ of the cervix) unless in complete remission and off therapy for a minimum of 3 years; active infection, or pregnancy.
Patients may have had treatment for no more than three prior relapses. Relapse is defined as progression following initial therapy (i.e. radiation ± chemo if that was used as initial therapy). The intent therefore is that patients had no more than four prior therapies (initial and treatment for three relapses). Patients with prior or current treatment with temozolomide were eligible provided they did not progress or were not progressing while on temozolomide.
The protocol was approved by the each institutional review board and conducted in accordance with institutional and federal guidelines for human investigation. All patients signed an approved informed consent form.
Stratification
Patients were stratified into two groups (Groups A and B) based on whether they are taking EIAEDs:
-
Group A: Patients who were not receiving EIAEDs.
-
Group B: Patients who were receiving EIAEDs.
EIAEDs included carbamazipine, oxycarbamazine, phenytoin, fosphenytoin, phenobarbital and primidone. All other anticonvulsants were considered non-EIAEDs.
Patients enrolled into Group B had to remain on EIAEDs throughout their treatment. If the EIAED had to be changed, another EIAED was used. If patients in Group A were required to start anticonvulsant medications, all efforts were made to use a non-EIAED. Any patients completing cycle 1 of therapy or patients removed for toxicity before cycle 1 was completed were evaluable for the primary endpoint. Patients removed from study before cycle 1 was completed (for reasons other than toxicity) were replaced.
Treatment
The starting dose of temozolomide was 150 mg/m2/day for 5 days every 28 days (one cycle); the dose could be increased in subsequent cycles to 200 mg/m2/day according to dosing guidelines described below. For cycle 1 only, temozolomide was started 7 days after the start of gefitinib.
Patients were treated with gefitinib on a continuous once daily dosing schedule, with 35 days constituting cycle 1, and 28-treatment days constituting subsequent treatment cycles. The dose escalation schema using the 250 mg formulation of the drug was as follows:
Dose levels | Dose (mg/day) |
|---|---|
−1 (starting dose level) | 250 |
1 | 500 |
2 | 750 |
3 | 1,000 |
4 | 1,250 |
5 | 1,500 |
6 | 2,000 |
7 | 2,500 |
Corticosteroids were used in the smallest dose to control symptoms of cerebral edema and mass effect, and discontinued if possible. Because of the potential for corticosteroids to induce CYP3A4, investigators were instructed to be especially alert for toxicity in patients weaned off corticosteroids during ongoing treatment with gefitinib. Anti-seizure medications were used as indicated. When clinically appropriate, the use of non-EIAEDs was encouraged. If during the course of the study, a change in AED was required or additional agents were required, patients on non-EIAEDs were not switched to an EIAED nor have an EIAED added to the regimen. Subjects who began treatment and required a change in drug were switched to another EIAED in order to remain on study, since change to a non-EIAED would place such subjects at risk for dose related toxicities. Concentrations of AEDs metabolized by CYP3A4 enzyme pathway were monitored at intervals to ensure that these remain within the therapeutic range.
Maximum tolerated dose and dose-limiting toxicity
The dose-limiting toxicity (DLT) was defined as follows (using the NCI-CTC version 2.0)
-
Any non-hematological grade-3 toxicity.
-
Any grade-4 hematological toxicity.
-
Protracted (greater than 2 weeks) grade-2 cardiac, pulmonary, renal, or CNS toxicities.
-
Toxicity that resulted in therapy interruption for greater than 2 weeks.
DLT was assessed in cycle 1. Cycle 1 started with gefitinib given alone on days 1–7; temozolomide was added on days 8–12. Gefitinib was continued on a daily basis. Cycle 2 began on day 35, or 28 days after the initial doses of temozolomide. Cycle 2 and all subsequent cycles were 28 days in length. Temozolomide was given on days 1–5 of cycle 2 and subsequent cycles. Dose escalation of gefitinib or temozolomide was not allowed in cycle 1. Assuming no DLTs or significant clinical deterioration after the first 35-day cycle, patients were treated with another cycle of gefitinib and temozolomide. Prior to cycle 3, patients underwent a clinical and radiographic tumor re-staging. As long as the tumor was stable in size or smaller and the patient was clinically stable or improved without dose-limiting toxicity, patients received additional cycles of gefitinib and temozolomide in 28-day cycles. Treatment was continued as long as there were no unacceptable toxicities (DLTs) and no evidence of tumor progression.
The MTD was based on the assessment of DLT during the first 35 days of treatment only (cycle 1), and was defined as the dose at which fewer then one-third of patients experience a DLT to gefitinib. The MTD was the dose level at which 0/3 or 1/6 patients experience DLT with the next higher dose having at least 2/3 or 2/6 patients encountering DLT. Toxicity was based upon toxicity felt secondary to gefitinib, rather than due to temozolomide. Three patients in Group A or Group B were treated at each dose level, and could be enrolled simultaneously. If one DLT was encountered, an additional three patients were added to that dose level. If at any point two DLTs were encountered within a given dose level, then the MTD had been exceeded and three more patients are treated at the next lower dose (if only three patients were previously treated at that prior dose).
Pharmacokinetic evaluation
Sample collection
The major pharmacokinetic goal of this trial was to determine the pharmacokinetic characteristics of gefitinib for brain tumor patients receiving concurrent therapy with EIAEDs. For all treated patients, serial blood samples at specified times were obtained before and after gefitinib administration on day 1 of the first treatment cycle. Heparinized blood specimens were collected at the following times during the first treatment cycle:
-
Day 1: Prior to drug administration, 30 min, 1, 2, 3, 4, 5, 8, 12 and 24 h after administration of the first day’s dose.
-
Days 8 and 29 (trough levels): Trough levels were obtained on days 8 and 29 of treatment by obtaining specimens immediately prior to the day’s oral dose.
Analytical method
Concentrations of gefitinib in plasma were analyzed by high performance liquid chromatography and quantified by mass spectrometric detection as described by Jones and Colleagues [6]. Analytical grade gefitinib and the deuterated (d8)–gefitinib internal standard (IS) were obtained from AstraZeneca Pharmaceutical (Macclesfied, UK). The analytical standard for the -O-desmethyl metabolite of gefitinib was not available, therefore not measured.
Gefitinib was isolated from plasma by liquid–liquid extraction. Briefly, 900 μl of plasma was spiked with 100 μl (50 ng) of the IS followed by the addition of 1 ml of 1 M sodium hydroxide and 6 ml of methl-tert butyl ether. After circular rotation for 1 h, the samples were centrifuged (4,000 rpm) at 25°C for 3 min. The organic layer was evaporated to dryness under a gentle stream of nitrogen in a 25°C water bath. The dry residue was reconstituted with 250 μl of mobile phase and sonicated for 5 min. Following re-centrifugation, 100 μl of sample was auto-injected at room temperature onto a reverse phase HPLC system (HP series II 1090 HPLC system, Hewlett Packard, Palo Alto, CA). The mobile phase consisted of 80% acetonitrile: 20% aqueous ammonium acetate (1% w/v) pumped at a flow rate of 1.0 ml/min. Separation of gefitinib was accomplished using an Inertsil ODS3 (3 μm, 2.1 1D × 150 mm; HiChrom Ltd., Berkshire, UK) column preceded by an Inertsil ODS3 cartridge guard column. Mass spectrometric (MS) detection was performed by a Finnigan LCQTM spectrometer, San Jose, CA, equipped with an atmospheric pressure chemical ionization (APCI) probe. The MS settings were: vaporizer temperature 450°C; sheath gas (N2) flow rate −59.75 arb; current 5.03 μA; voltage 4.17 kV; capillary temperature 176°C and capillary voltage 3 kV.
In the MS/MS mode, the collision energy was 52%. For peak identification, full-scan mass spectra were acquired in a positive ion mode. The MS/MS scan range for gefitinib was 120–500. Selected ion monitoring was used for the determination of the ammonium adducts [M + NH4] and the compound’s respective fragment ion: gefitinib (m/z 447.2–136.2).
Data acquisition and integration of the chromatograms were performed using XcaliberTM LCQUAN program (Finnigan, San Jose, CA). The chromatographic data were analyzed by linear least-squares regression with a weighting factors of 1/x 2 generating an eight-point calibration curve of area ratios for gefitinib. The calibration curves were linear (R 2 > 0.99) over the range from 0.5 to 500 ng/ml. Concentrations above the highest calibration standard were diluted into the linear range of the calibration curve. The slope of seven separate calibration curves used in the analysis of samples over a two-year span ranged from 0.007 to 0.010 with a mean valve of 0.009 ± 0.0018 (SD). Samples were repeated if the independent quality control (QC) samples at the low (1.5 ng/ml) exceeded the theoretical value by 20% and the medium (40 ng/ml) or high (90 ng/ml) QC by 15%. Of the 28 analytical runs performed, only two of the duplicate QCs failed. The interday precision for gefitinib was 10.79, 8.91, and 8.29% for the low, medium and high QC samples.
Pharmacokinetic analyses
Gefitinib plasma concentrations were analyzed by non-compartmental methods. The time interval relative to the administration of gefitinib and the actual sample times were used for the determination of the time to peak (t max) and the estimation of the area under the plasma concentration-time curve (AUC0–24) by linear trapezoidal rule. Peak concentrations (Cpmax) were determined by inspection of each individual’s plasma concentration–time curve. Since the number of patients per dose level was relatively small in this dose-finding study, the AUC, Cpmax and trough levels were dose-normalized for statistical comparisons.
Statistical considerations for PK analyses
Pharmacokinetic parameters are reported as mean values ±SD. The relationship between gefitinib dose administered (mg) and AUC0–t were analyzed by Spearman’s correlation coefficient and linear regression analysis. Differences between the two groups with respect to the kinetic variables were evaluated using an unpaired 2-tailed (t) test. Two-tailed probability values of less than 0.05 were regarded as statistically significant.
Dose modifications for subsequent cycles of gefitinib
The most common non-hematologic toxicities in prior studies of gefitinib have been skin rash and diarrhea. Tolerable grade-2 diarrhea and skin rash did not require temporary discontinuation of treatment as these toxicities could improve despite continued treatment with gefitinib. Symptomatic treatment of diarrhea with immodium or other antidiarrheals was recommended. At any time during any treatment cycle, grade-2 skin rashes and diarrhea that were intolerable or unacceptable to the patient, gefitinib was temporarily held until resolution of the toxicity to grade 1 or less (grades 0–1) and subsequently re-started at the same dose. If symptomatic grade-2 diarrhea and skin rash recurred after re-instituting treatment at the initial dose and again required temporary discontinuation, treatment was held until resolution to grade 1 or less (grades 0–1) and re-instituted at a dose 250 mg less than the initial dose. For grade-2 non-hematological toxicity that was medically concerning [e.g. prolonged (greater than 2 weeks) cardiac, pulmonary or neurotoxicity], treatment was held until resolution and re-instituted at a dose 250 mg less than the initial dose. For Grades 3 and 4 toxicities, treatment was discontinued and patients re-evaluated at least weekly until resolution to grade 1 or less (grades 0–1). Treatment was reinstituted at a dose of 250 mg less than the previous daily dose. Clinical neurological events that could be due either to drug toxicity, treatment effect (e.g. radiation injury) or tumor progression were resolved by MR imaging.
Dosing guidelines for temozolomide
Patients receiving the combination of temozolomide plus gefitinib started temozolomide at 150 mg/m2/day for five consecutive days in the first cycle. For the first cycle, temozolomide began on day 7 after the start date of gefitinib. For patients already on temozolomide, gefitinib treatment began 7 days before further temozolomide therapy. Every month of therapy was considered a cycle after cycle 1 was complete. Temozolomide was given on days 1–5 of cycle 2 and subsequent cycles. If there was no toxicity during the initial temozolomide cycle, patients could be treated at 200 mg/m2/day for subsequent cycles. Treatment cycles were repeated every 28 days following the first daily dose of temozolomide from the previous cycle. Temozolomide was not escalated beyond 200 mg/m2/day.
The dose of temozolomide administered for subsequent cycles was determined according to the nadir ANC and nadir platelet count of the immediately previous cycle for temozolomide (see the following table).
Dose–adjustment criteria | |||
|---|---|---|---|
Nadir toxicity level | Nadir ANC/μl | Nadir platelets/μl | Temozolomide modification |
0 | ≥2,000 | ≥100,000 | Dose unchanged from previous |
1 | 1,500–1,999 | 75–99,999 | Dose unchanged from previous |
2 | 1,000–1,499 | 50–74,999 | Dose unchanged from previous |
3 | 500–999 | 25–49,999 | Decrease dose to next lower dose level |
4 | <500 | <25,000 | Decrease dose to next lower dose level |
Non-Hematologic Criteria: Subsequent courses could start (as long as the treatment was beneficial) after complete resolution of toxicities to Grade 2 or less. A minimum 2-week rest period was required if there was Grade 3 or greater non-hematologic toxicity. Dosages for the subsequent course were one dose level below the dose that produced toxicity of Grade 3 or greater as follows:
Dose level | −2 | −1 | 0 |
Temozolomide (5 days) | 100 mg/m2 | 150 mg/m2 | 200 mg/m2 |
If the patient experienced Grade 3 or greater hematologic or non-hematologic toxicity with a dose of 100 mg/m2 temozolomide, he or she was discontinued from the study.
Pre-treatment evaluations
A complete history and neurological examination, as well as documentation of evaluable disease were performed on all patients. Baseline scan were obtained within 14 days of registration and on a steroid dosage that has been stable for 5 days. All patients had a baseline ophthalmologic examination performed prior to beginning drug administration within 21 days of registration. Pre-study laboratory tests included EKG, CBC, platelets, PT, PTT, serum electrolytes, calcium, phosphorus, magnesium, albumin, alkaline phosphatase, serum creatinine, creatinine clearance or estimated creatinine clearance (140-age × weight in kg/72 × crserum) × 0.85 for females, bilirubin, SGPT, SGOT, and serum pregnancy test for women of childbearing potential. Pre-study laboratory tests were obtained with 14 days of registration.
Evaluations during study
CBC and platelets were performed every week during the treatment; PT, PTT, serum electrolytes, calcium, phosphorus, magnesium, albumin, alkaline phosphatase, serum creatinine, bilirubin, SGPT, SGOT, bilirubin, BUN were performed every 4 weeks prior to each cycle. A brain MRI was done prior to every other cycle. Patients with stable or responding disease could continue treatment, assuming no dose-limiting toxicity, until tumor progression. Tumor progression was determined by the use of sequential MR imaging, and was defined as an enlargement of the tumor cross-sectional enhancement area by 25%. Complete or partial response required complete resolution of enhancement or a greater than 50% decrease in tumor area, respectively; stable disease required no change, or less than 25% increase or decrease in tumor area. All responses required that there was no change or a decrease in steroids and patients had to have clinically stable disease.
Criteria for removal from study
Patients were removed from the study due to disease progression, unacceptable toxicity, prolonged treatment delays, or medical or psychiatric illness which in the investigator’s judgment rendered the patient incapable of further therapy. In addition, patients could withdraw from the study at any time for any reason.
Results
There were 28 patients registered from seven institutions. Of these, two were not evaluable. One had an ineligible pathology on central review, and one patient declined treatment after registration. The results include information from 26 eligible and evaluable patients. Patient characteristics are shown in Table 1. Ten patients were enrolled in Group A (no EIAED use) and 16 in Group B. The median age was 54 years in Group A and 48 years in Group B. The most common diagnosis was GBM.
The DLT for patients not on EIAEDs was elevation of liver function studies, including AST and ALT, when treated with gefitinib at a dose of 500 mg/day. Two patients out of six had this event. Transaminase elevations typically resolved within 2–4 weeks for grade-3 or 4 toxicities. Only one patient required longer than 4 weeks for resolution. At a dose of 250 mg/day dose level of gefitinib, only one patient out of six had dose-limiting diarrhea. The MTD for gefitinib in this patient group is 250 mg/day when used with temozolomide. The DLT for patients on EIAEDs was diarrhea, nausea and vomiting. Two patients out of three had this event at a dose of 1,250 mg/day. No DLTs were noted in six patients treated at a dose of 1,000 mg/day, the MTD. Tables 2 and 3 describe all grades 3 and 4 toxicities of the treatment for Groups A and B. Of note, rash was only a minor problem in this study. Only one grade-3 rash was recorded in a patient in Group A. The remainder of the patients had only grade 1 or 2 rash. Grade 3 diarrhea was not common in either group, but was seen more often in Group B patients. Hematological toxicity was not significant, and was typical for temozolomide when used as a single agent.
Pharmacokinetic results
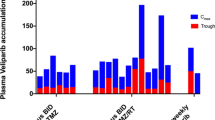
The mean (±SD) pharmacokinetic parameters for gefitinib are summarized in Table 4. Comparison of the dose-normalized pharmacokinetic parameters between the two groups (non-EIAEDs versus EIAEDs) indicates an apparent effect of EIAEDs on the systemic disposition of gefitinib (Table 5). The time to peak concentrations were similar for both groups. Mean peak plasma concentrations were 1.5-fold lower in the EIAED group, compared to the non-EIAED group. More importantly, the systemic exposure (AUC) to gefitinib was significantly (P = 8.0 × 10−5) lower in the EIAED group. Trough levels on days 1, 8 and 29 were all significantly lower for the EIAED group. Trough levels increased in both groups consistent with its reported terminal half-life (21–51 h) achieving steady-state concentrations at day 8. There was no statistical difference between the days 8 and 29 trough levels in either group.
Discussion
The overall metabolism of gefitinib is primary regulated by the abundant, highly inducible CYP3A4/5 [12]. However, the non-inducible, highly polymorphic CYP2D6 is also responsible for the metabolism of gefitinib to one of its major metabolite, desmethy-gefitinib [12]. We observed that patients receiving EIAED has significantly lower peak concentrations and exposure (AUC0–24) to gefitinib compared to the non-EIAED patients. This observation is consistent with another recent report [16] and our NABTC 00-01 phase-1 single-dose trial with gefitinib alone in patients with recurrent malignant gliomas [10]. In the NABTC 00-01 trial, patients on EIAEDs received escalating doses of gefitinib ranging from 500 to 2,000 mg daily. The MTD for patients receiving EIAEDs was 1,250 mg. The EIAED affects on the pharmacokinetic parameters of gefitinib observed in the NABTC 00-01 trial are similar to those observed in the current combination trial (Table 5). Our trial was not designed to detect a pharmacokinetic interaction between either gefitinib or temozolomide. However, we did not expect that gefitinib would affect the pharmacokinetics of temozolomide, since temozolomide is metabolized by non-enzymatic, chemical degradation process [1]. However, a modest pharmacokinetic interaction between another EGFR tyrosine kinase inhibitor, erlotinib which is also a CYP3A4 substrate and temozolomide has been reported [15].
Somatic mutations in the EGFR gene have been identified which enhance the activity of gefitinib in lung tumors [11, 14]. Indeed, there are reports of patients with brain metastases from non-small-cell lung cancer responding to gefitinib [4]. Of note, gefitinib inhibits p-glycoprotein (pgp/ABCB1) as well as, the breast cancer resistance protein (BCRP/ABCG2) both of which are components of the capillary endothelial cell of the brain [2, 7]. The mutant EGFR receptors are inhibited by gefitinib concentrations of 0.2 μmol/l (89 ng/ml), whereas, the wild-type EGFR receptor requires concentrations of 2 μmol/l (894 ng/ml) for complete inhibition [13]. Mean steady-state trough plasma concentrations (day 8 or day 29) of gefitinib ≥2 μmol/l were not achieved in either of our patient groups (Table 4). However, preliminary data obtained from our NABTC 00-01 trial suggests a preferential distribution of gefitinib from blood into brain tumor tissue. Pre-surgical treatment of non-EIAED patients with gefitinib 500 mg/day for 7 days resulted in gefitinib concentrations in brain tumor tissue 18 times higher than plasma (unpublished). The tumor concentrations approached or exceeded the concentration (2 μmol/l) reportedly necessary for wild-type EGFR inhibition. Inhibition of EGFR phosporylation was evident in these patient’s tumors by comparison of the tissue prior to resection versus post gefitinib treatment.
In conclusion, the combination of gefitinib and temozolomide is tolerable and the recommended starting phase-2 dosing criteria for patients on or not on EIAEDs are suggested. It should be emphasized that the DLT was evaluated in the first cycle of therapy only, in patients receiving gefitinib in combination with temozolomide at a dose of 150 mg/m2/day for 5 days. Using temozolomide at a dose of 200 mg/m2/day in combination with gefitinib was possible in some patients but was not the intent of this study. Future studies should be considered using this combination in patients with newly diagnosed disease, with the potential to increase efficacy over that seen with temozolomide alone in this patient population.
References
Baker SD, Wirth M, Statkevich P, Reidenberg P, Alton K, Sartorius SE, Dugan M, Cutler D, Batra V, Grochow LB, Donehower RC, Rowinsky EK (1999) Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin Cancer Res 5:309–317
Begley DJ (2004) Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol Ther 104:29–45
Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, Bigner DD (1990) Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res 50:8017–8022
Cappuzzo F, Calandri C, Bartolini S, Crino L (2003) ZD 1839 in patients with brain metastases from non-small-cell lung cancer (NSCLC): report of four cases. Br J Cancer 89:246–247
Chakravarti A, Dicker A, Mehta M (2004) The contribution of epidermal growth factor receptor (EGFR) signaling pathway to radioresistance in human gliomas: a review of preclinical and correlative clinical data. Int J Radiat Oncol Biol Phys 58:927–931
Jones HK, Stafford LE, Swaisland HC, Payne R (2002) A sensitive assay for ZD1839 (Iressa) in human plasma by liquid-liquid extraction and high performance liquid chromatography with mass spectrometric detection: validation and use in Phase I clinical trials. J Pharm Biomed Anal 29:221–218
Kitazaki T, Oka M, Nakamura Y, Tsurutani J, Doi S, Yasunaga M, Takemura M, Yabuuchi H, Soda H, Kohno S (2005) Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 49:337–343
Lassman AB, Rossi MR, Raizer JJ, Abrey LE, Lieberman FS, Grefe CN, Lamborn K, Pao W, Shih AH, Kuhn JG, Wilson R, Nowak NJ, Cowell JK, DeAngelis LM, Wen P, Gilbert MR, Chang S, Yung WA, Prados M, Holland EC (2005) Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res 11:7841–7850
Leuraud P, Taillandier L, Medioni J, Aguirre-Cruz L, Criniere E, Marie Y, Kujas M, Golmard JL, Duprez A, Delattre JY, Sanson M, Poupon MF (2004) Distinct responses of xenografted gliomas to different alkylating agents are related to histology and genetic alterations. Cancer Res 64:4648–4653
Lieberman FS, Cloughesy T, Fine H, Kuhn J, Lamborn K, Malkin M, Robbins HI, Yung WA, Wen P, Prados M (2004) NABTC phase I/II trial of ZD-1839 for recurrent malignant gliomas and unresectable meningiomas. J Clin Oncol 2004 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 22:108s [Abstract 1510]
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139
McKillop D, McCormick AD, Millar A, Miles GS, Phillips PJ, Hutchison M (2005) Cytochrome P450-dependent metabolism of gefitinib. Xenobiotica 35:39–50
McKillop D, Partridge EA, Kemp JV, Spence MP, Kendrew J, Barnett S, Wood PG, Giles PB, Patterson AB, Bichat F, Guilbaud N, Stephens TC (2005) Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther 4:641–649
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Prados MD, Lamborn KR, Chang S, Burton E, Butowski N, Malec M, Kapadia A, Rabbitt J, Page MS, Fedoroff A, Xie D, Kelley SK (2006) Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neurooncology 8:67–78
Reardon DA, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH, Desjardins A, Sathornsumetee S, Herndon JE 2nd, Dowell JM, McLendon RE, Provenzale JM, Sampson JH, Smith RP, Swaisland AJ, Ochs JS, Lyons P, Tourt-Uhlig S, Bigner DD, Friedman HS, Rich JN (2006) Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin Cancer Res 12:860–868
Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, Kao JC, Stenzel TT, Ahmed Rasheed BK, Tourt-Uhlig SE, Herndon JE 2nd, Vredenburgh JJ, Sampson JH, Friedman AH, Bigner DD, Friedman HS (2004) Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol 22:133–142
Stark AM, Witzel P, Strege RJ, Hugo HH, Mehdorn HM (2003) p53, mdm2, EGFR, and msh2 expression in paired initial and recurrent glioblastoma multiforme. J Neurol Neurosurg Psychiatry 74:779–783
Stea B, Falsey R, Kislin K, Patel J, Glanzberg H, Carey S, Ambrad AA, Meuillet EJ, Martinez JD (2003) Time and dose-dependent radiosensitization of the glioblastoma multiforme U251 cells by the EGF receptor tyrosine kinase inhibitor ZD1839 (‘Iressa’). Cancer Lett 202:43–51
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Acknowledgments
This study was supported by the following grants: NABTC grants: CA62399, CA62422, CA62412, U01CA62407-08, CA62455-08, U01CA62405, CA62426, U01CA62399 No. 022330 (for NABTC98-03 only), U01CA62399, 5-U01CA62399-09, U01CA62399-09, and U01CA62421-08. GCRC grants: M01-RR00079, CA16672, M01-RR00633, M01-RR00056, M01-RR0865, M01-RR00042, M01-RR03186.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prados, M.D., Yung, W.K.A., Wen, P.Y. et al. Phase-1 trial of gefitinib and temozolomide in patients with malignant glioma: a North American brain tumor consortium study. Cancer Chemother Pharmacol 61, 1059–1067 (2008). https://doi.org/10.1007/s00280-007-0556-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-007-0556-y