
Abstract
Purpose: The aims of the study were (a) to characterise the pharmacokinetics (PK), including inter-individual variability (IIV) and inter-occasion variability (IOV) as well as covariate relationships and (b) to characterise the relationship between the PK and the haematological toxicity of the component drugs of the fluorouracil (5-FU)—epirubicin (EPI)—cyclophosphamide (CP) regimen in breast cancer patients. Patients and methods: Data from 140 breast cancer patients, either within one of different studies or in routine clinical management, were included in the analyses. The patients were all treated with the fluorouracil-epirubicin-cyclophosphamide (FEC) regimen every third week for 3–12 courses, either in standard doses, i.e. 600/60/600 mg/m2 of 5-FU, EPI and CP, respectively, or according to a dose escalation/reduction protocol (tailored dosing). PK data were available from 84 of the patients, whereas time-courses of haematological toxicity were available from 87 patients. The data analysis was carried out using mixed effects models within the NONMEM program. Results: The PK of 5-FU, EPI and 4-hydroxy-cyclophosphamide (4-OHCP), the active metabolite of CP, were described with a one-compartment model with saturable elimination, a three-compartment linear model and a two-compartment linear model, respectively. No clinical significant correlation was found between PK across drugs. The unexplained variability in clearance was found to be less within patients, between courses (inter-occasion variability, IOV) than between patients (inter-individual variability, IIV) for EPI and 5-FU. For 4-OHCP, however, the IIV diminished by approximately 45% when significant covariates were included and the final population model predicts an IIV that is equal to IOV. Significant covariates for elimination capacity parameters were serum albumin (5-FU, EPI and 4-OHCP), creatinine clearance (5-FU), bilirubin (EPI) and body surface area (BSA) (4-OHCP). Elimination capacity of 5-FU and EPI was not related to BSA and for none of the studied drugs did body weight explain the PK variability. The time-course of haematological toxicity after treatment was well described by a semi-physiological model that assumes additive haematological toxicity between CP and EPI with negligible contribution from 5-FU. The influence of G-CSF could be incorporated into the model in a mechanistic manner as shortening the maturation time to 43% of the normal duration and increasing the mitotic activity to 269% of normal activity. Conclusions: The models presented describe the dose-concentration-toxicity relationships for the FEC therapy and may provide a basis for implementation and comparison of different individualisation strategies based on covariates, therapeutic drug monitoring and/or pharmacodynamic (PD) feedback. The PD model extends on previous semi-mechanistic models in that it also takes G-CSF administration into account.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chemotherapy of breast cancer is frequently carried out using drug combinations, which have been found superior to single drug treatment [13]. The 5-fluorouracil-epirubicin-cyclophosphamide (FEC) regimen is a widely established regimen in the treatment of breast cancer. The standard dosing is 600/60/600 mg/m2 for 5-fluorouracil (FU)/epirubicin (EPI)/cyclophosphamide (CP) every third week. More dose intensive schedules have however been used [2, 3, 28, 36, 46, 47]. Haematological toxicity is the shared dose limiting acute event for the three drugs and granulocyte colony stimulating factors (G-CSF) have occasionally been included in the treatment allowing increased dose intensity [2, 47].
Although the main reason for the lack of distinct dose-response and dose-toxicity relationships for anti-cancer agents is variability in pharmacodynamics (PD), the variability in the pharmacokinetics (PK) of an anti-cancer agent among individuals (inter-individual variability, IIV), within one individual from one course to another (inter-occasion variability, IOV) and, in case of multi-therapies, drug–drug interactions, may contribute as well. That the PK can be used as a biological marker for anti-tumoral effect has been shown for methotrexate and teniposide, among others [10, 38]. Hence, describing and quantifying the PK of all component drugs of a regimen, including variability and, if possible, explaining the variability, and in addition describing relationships between PK and dose-limiting side-effects, are of clear importance in the process of developing dosing strategies for anti-cancer regimens. Despite that co-variation in the PK between component drugs within regimens cannot be presumed, studies where all component drugs within multiple-regimens are simultaneously monitored, are rare in the literature [10, 14, 33, 40].
We previously reported the IIV and IOV in the PK parameters of the component drugs of the FEC regimen, studied in 21 breast cancer patients [40]. The results indicated a large variability, particularly for the active metabolite of CP, 4-hydroxycyclophosphamide (4-OHCP), in both IIV and IOV. The IOV of all three studied drugs were found to be less than the corresponding IIV. Furthermore, no significant correlation was found between any of the PK parameters of the component drugs. However, the limited number of patients in the study precluded confident conclusions.
Although the benefit of increasing dose intensity in chemotherapy of breast cancer has been questioned [22], it is expected that the higher the dose given to a patient, the higher the probability of achieving effective exposure of the tumour [6]. However, the risk of obtaining fatal toxicity as a consequence of the treatment makes the clinicians uncertain about how to optimally dose the individual patient. Hence, despite lack of knowledge in which is the effective concentration/exposure for a drug, therapeutic drug monitoring might be worthwhile, provided that the relationships between the PK and the dose-limiting toxicities are known. However, the lack of models derived from the anti-cancer multi-therapy situation in which the component drugs frequently show overlapping toxicities, has so far precluded the benefit of therapeutic drug monitoring (TDM) in the majority of anti-cancer regimens.
The aims of the study were (a) to characterise the PK, including IIV and IOV as well as covariate relationships and (b) to characterise the relationship between the PK and the haematological toxicity of the component drugs of the FU-EPI-CP regimen in breast cancer patients.
Patients and methods
Patients and treatment
Data from 140 breast cancer patients, treated during the years 1992–1997, either within one of different studies or within the routine medical service, were included in the analysis (Table 1). All study protocols were approved by the Local Ethics Committee; study patients, pilot patients and patients treated in clinical routine gave their consent for the PK sampling. The patients were all treated with the FEC regimen every third week at 3–12 occasions, either in standard doses, i.e. 600/60/600 mg/m2 of 5-FU, EPI and CP, respectively, or according to a dose escalation/reduction protocol (tailored dosing). The initial doses administered to the patients who were treated with tailored FEC were 600/75/900 mg/m2. The following doses were either escalated stepwise up to a maximum of 600/120/1,800 mg/m2 or decreased to a minimum of 300/38/450 mg/m2, based on the nadir and dosing day leukocyte and thrombocyte count. All patients received in addition tropisetron and betamethasone.
Group I
Twenty-one patients were treated with standard FEC. PK samples were withdrawn at 2–7 different time points after start of drug treatment at one or several occasions. Leukocyte counts (WBC) were made in association with the treatment days only. The PK in this group of patients has been reported previously [40].
Group II
Thirty-two patients were treated with either standard FEC or tailored FEC every third week. At 2–4 different time points (10 min to 24 h) after the cytotoxic drug administration, in association with one or several dosing occasions, blood samples were taken for determination of drug concentration. The haematological toxicity was measured at dosing days only.
Group III
Thirty-five patients were treated with tailored FEC every third week. Blood samples were taken, according to the design used in Group II, for determination of drug concentration at one or several occasions. Blood samples for leukocyte count were withdrawn at day 1, the day of treatment, and thereafter at day 7, 10/11, 14/15 and in addition in association with dosing days.
Group IV
Fifty-two patients were within the routine medical service treated either with standard or tailored FEC. No granulocyte stimulating factors were given to those patients. Routine blood samples for leukocyte count were in general taken at the 7, 10/11 and14/15 days after and in association with the treatment days. No PK information was available. The data from this group of patients was retrospectively collected from the patient records.
All patients who received more than 1,000 mg/m2 CP, were in order to protect the bladder treated with mesna, supplied as Uromitexan®(ASTA Medica, Täby, Sweden), in the doses that were 20% of the CP dose. Mesna was administered immediately before start of FEC therapy and thereafter at 4–6 and 8–10 h in relation to the CP dosing. Approximately 70% of the patients (group III, see below) that received tailored FEC were, in order to be able to increase dosing intensity, treated with G-CSF. The choice of G-CSF dosing strategy was determined by the disease. Patients that suffered from metastatic disease, defined as stage IV disease according to IUCC, were given 13.4 million units daily, supplied as Granocyt® (Rhone-Poulenc Rorer, Helsingborg, Sweden/Aventis, Stockholm, Sweden) during 10 days, starting 2 days after the FEC therapy. The remaining patients received G-CSF, supplied as Neupogen® (Amgen, Bromma, Sweden), 0.5 million units/kg total body weight once daily during 2 weeks starting 2 days after FEC treatment. All patients in group III were also given ciprofloxacin, supplied as Ciproxin® (Bayer Sverige AB, Göteborg, Sverige) 500 mg twice daily during 2 weeks, starting 2 days after the FEC treatment.
The majority of the courses were administered starting with a 15-min infusion of CP, followed by 5-FU given as an intravenous bolus dose and EPI given either as a bolus or as a 1-h infusion. The following covariates were recorded in association with the treatment days at one or several dosing occasions: age, weight (WT), height (HT), body surface area (BSA), serum aspartate transaminase (AST) and serum alanine transaminase (ALT), serum bilirubin (BILI), serum albumin (ALB), serum creatinine (Cr) and creatinine clearance (CLCR). CLCR was calculated according to Cockroft and Gault [4].
Sampling for pharmacokinetics
Sampling strategy
Samples were withdrawn within four different sampling windows in order to cover the main part of the concentration-time profile for each drug. The first sample was taken approximately 10 min after administration of 5-FU whereas the second, third and fourth sample was withdrawn around 1, 6 and 15–24 h, respectively, after start of therapy. Thus, the first sample was often collected during the EPI infusion.
Handling of samples
The blood samples were collected in EDTA-coated Vacutainer tubes (Becton-Dickinson, Plymouth, UK), immediately put on ice water bath and thereafter, within 15 min, centrifuged at 4°C to obtain plasma which was immediately put in -70° until analysis. Plasma for determination of 4-OHCP was treated as has been previously recommended [40].
The chemical assays
The methods used to determine the drug concentrations have been described elsewhere [40, 41].
Pharmacokinetic analysis
The data collected from groups I, II and III were included in the PK analysis that was carried out using mixed effects modelling within the program NONMEM version VI beta using the FOCE method with interaction [1]. The search for appropriate models was guided by graphical evaluation within the program Xpose, as well as the, by NONMEM estimated, objective function which is a measurement of the goodness of fit. NONMEM also produces Bayesian individual parameter estimates. Individual model predicted concentrations are based on those estimates. The significance level required to keep a parameter in a hierarchical model was set to p<0.01, which corresponds to a drop in the objective function by 6.63. First, the structural and statistical models were developed, including inter-individual (IIV) and inter-occasion (IOV) variability terms, and second the covariates were added to the respective model using an automated model building procedure [24]. Candidate covariates were all those monitored (See Patients and treatment). Missing covariates were in the model building process replaced with the respective median value. IOV was introduced into the models as suggested previously [25]. Proportional and slope-intercept residual error models were evaluated for all three datasets.
Covariate relationships were considered only on parameters on which IIV and/or IOV were significant. The automated model building procedure used works stepwise, considering both linear and non-linear relationships between each candidate covariate and the PK parameters. In the forward search each covariate is in each step included into the model separately. The most significant model is kept in the next step and into that model each of the remaining covariates is again included, one at a time. This continues until no additional covariate is judged significant. Thereafter follows the backward search in which the included covariates are excluded one at a time, again to test their significance in the model. The significance levels used in the present analysis were p<0.05 in the forward search, whereas a higher significance level, p<0.01, was used in the backward search. Linear and non-linear (piecewise linear splines) models were evaluated for all covariates. In order to identify any sensitivity of the final models for individuals that alone are responsible for (driving) a covariate relationship each model was finally re-run ten times using only nine-tenth of each data set at a time (Jack knife).
Pharmacokinetic-pharmacodynamic analysis
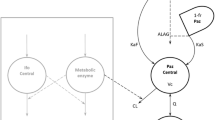
A model describing the time course of the haematological toxicity after single drug administration proposed previously [15] was used in the PK–PD analysis with the exception that a combined effect of more than one drug was considered in the present analysis. The model consists of five compartments of which one is a compartment containing proliferating cells. That compartment is linked to the white blood cell compartment via a maturation chain consisting of three compartments (Fig. 1).

The semi-physiological model used in the PK/PD analysis. MTT is the mean transit time that represents the maturation chain; k prol, the prolongation rate constant; k circ, the elimination rate constant for circulating leukocytes and k MTT, the maturation rate constant [WBCbase/WBC(t)]γ, is the feedback component where WBCbase is the predicted baseline WBC and WBC(t), the predicted WBC concentration at time t. C DRUG(t) and SDRUG are the drug concentrations and pharmacodynamic slope factors, respectively
The model is described by the physiological parameters WBC baseline (WBCbase), i.e. the concentration of leucocytes in the absence of any drug therapy, mean transit time (MTT), which is the average time it takes a cell to mature and appear in the circulating blood, and the gamma (γ) which is part of the feedback model [WBCbase/WBC(t)]γ. The feedback model imitates the endogenous colony stimulation factor feedback system that responds to changes in circulating white blood cell concentration. The first-order transfer between maturation compartments are given by the constant, k MTT, which in this model is equal to 4/MTT. The proliferation rate constant (k prol), is arbitrarily set to the value of k MTT, whereas the rate constant for disappearance of leukocytes from the circulation (k circ), was set equal to k MTT as previous investigations have showed that this type of data contain so little information about k circ that such an approximation is not detrimental.
The drug effect in the model acts on the proliferating rate constant and is a function of the drug concentration in plasma and a drug specific linear slope, such that the changes in the concentration of proliferative cells (Prol) are given by:
The drug concentration-time profiles were in the PD analysis fixed to values given by the typical individual parameters (group IV), or to the individual parameters obtained as empirical Bayes estimates (group III) from the PK analysis. Since only pre-dose observations of WBC were available from the patients in groups I and II, the analysis was based solely on the data from groups III and IV.
The analysis was carried out in two steps. In the first step, a model was created for data from patients from group IV, i.e. to which no G-CSF was administered. PK was for those patients assumed to be given by the typical individual parameters of the population model developed on the data from groups I and II. The slope for EPI (S EPI) was fixed to 17.5 L/mg, a value obtained under development of a corresponding PK/PD model in a previous study [40], whereas other system-specific and drug-specific parameters were estimated. In the second step, a model was developed to describe data from patients in group III. In this analysis, the concentration-time profiles were fixed to those provided by individual-specific parameters of the final PK model.
The PD modelling proceeded as follows. In all models, WBCbase, interindividual variability in WBCbase, and residual error were estimated. All other parameters were fixed to the values obtained in step one, unless otherwise specified. It was assumed that G-CSF could influence one or several of the following parameters k prol, MTT, k circ and γ. In addition, amplification in the maturation chain or an additive effect resulting from mobilisation of new Prol cells were tested for improvement of the model. The former was achieved by allowing the input into each compartment in the maturation chain to be equal to the disappearance from the previous compartment raised to an estimated exponent. The latter was achieved by an estimated additive factor on the right-hand side of Eq. 1. Thus, six types of possible effects were tested for their potential to describe data from group III. For each of these effects, it was investigated whether there may be a time-dependence in the effect of G-CSF. This was accomplished by allowing each estimated component to take on different values in the following three periods: the first half of each G-CSF course, the second half of each G-CSF course and in the post-G-CSF periods, before the start of a new course. The analysis proceeded by first assessing the fit of the model to the data when none or only one component were assumed to be influenced by G-CSF. Thereafter, combinations of effects and various overall time-effects on k prol or WBCbase were investigated. Finally, redundant parameters were omitted and the statistical significance of each component was assessed by sequential deletion from the final model and observing the increase in the objective function value. The FO method in NONMEM was used for the PD analyses and partly therefore a more conservative significance level, p<0.01, was required to keep a parameter in the final model.
Results
Pharmacokinetics
The covariate data collected are given in Table 1. Data from a total of 188 courses in 84 individuals was available for the PK analysis. The PK information on all three component drugs were available from 146 courses in 70 patients. The raw data for 5-FU, EPI and 4-OHCP are displayed in Fig. 2a, e and i. The final basic and covariate models are presented in Table 2. In addition, the respective basic/covariate population model and individual model predictions versus observations are apparent in Fig. 2b–d, f–h and j–l. Proportional error models described the residual error as well as did slope-intercept models in the data sets analysed.

Observed concentrations versus time for 5-FU (a), EPI (e) and 4-OHCP (i). Basic model predicted concentrations versus observed concentrations (b, f and J). Final covariate model predicted concentrations versus observed concentrations (c, g and k) and individual model predicted concentrations versus observed concentrations (d, h and l). Included are also lines of indentity
5-Fluorouracil
A total number of 327 observations from 162 occasions in 77 individuals was available in the PK analysis (Fig. 2a) A one-compartment model with saturable elimination was sufficient in describing the 5-FU data. The subroutine ADVAN 10 TRANS 1 was used in the analysis. IIV was significant on the parameters maximum elimination rate (V max) and volume of distribution (V). Introducing IOV on V max and the concentration at which elimination rate is half of V max (Km) further improved the fit. A strong correlation of 93% was found between the IOV of V max and Km. Simulations in Excel [30] showed that the IIV and IOV in CL at the plasma concentration 5 mg/L were 20 and 15%, respectively. Three individuals differed considerably from the rest of the population in their 5-FU PK. They were therefore excluded in the main analysis and analysed separately. The CL at 5 mg/L was in that group of patients estimated to 36.3 L/h. The candidate covariates ALB, BSA and CLCR were judged significant since they lead to a decrease in the objective function by 8.4, 9.1 and 10.8 units, respectively. Since values for ALB, BSA and CLCR was lacking from 23, 8 and 17% of the individuals the final covariate model was re-run with data from only the individuals from whom each covariate was known. The estimates from those runs did not differ from those obtained in the final covariate model. No strongly influential individuals were identified.
Epirubicin
A total of 544 observations, 177 occasions in 79 individuals, were available in the PK analysis (Fig. 1e). A three-compartment model was as expected from previous studies superior to a two-compartment model [34, 40]. The IIV was significant on CL and central volume of distribution (V1). However, when IOV was introduced on those parameters, it turned out that IIV could not be separated from IOV on V1. The IOV on CL was estimated to 7%. However, that parameter did not improve the fit significantly and was therefore not kept in the model (Table 2). No significant covariate relationships were found for V1 whereas introducing BILI and ALB as influencing factors on CL resulted in a decrease in the objective function of 10.0 and 8.6, respectively. Deleting data from individuals from whom covariates were missing (ALB 23%; BILI 14%) did not change the covariate effect estimates. The jack-knife procedure did not point at any influential individual. The final covariate model is specified in Table 2.
4-Hydroxy-cyclophosphamide
Data from 76 individuals, 548 observations from 171 occasions, was available in the analysis (Fig. 1i). Although 4-OHCP is a metabolite, models describing initially ascending concentrations over time did not describe the data significantly better than did a model describing concentration-time profile as following intravenous bolus administration. The best structural model was a linear two-compartment model (Table 2). The data supported IIV and IOV on the apparent CL (CL/fm) and V1/fm, but only IOV on V2/fm. A strong correlation (89%) was found between CL/fm and V/fm). The covariates ALB and BSA were significantly correlated to CL, whereas ALT and WT co-varied with the volume terms. Since one individual was found to strongly influence the relationship between CL and the covariates ALB and BSA, that individual was excluded from the dataset and the covariate relationships where re-evaluated. The final covariate model is hence not dependent on the influential individual (Table 2). The exclusion of ALB, ALT, BSA and WT caused an increase in the objective value of 16, 16, 17 and 25, respectively. Values for ALB, ALT, BSA and WT was lacking from 22, 13, 7 and 4% of the individuals. The estimates obtained from runs that did not include the individuals from whom the covariate values were unknown resulted in similar parameter estimates as did the final model.
To make certain that the PK did not vary between and within individuals due to either mesna treatment or grade of disease, the influence of mesna and metastasised disease were evaluated on the PK parameters in the final covariate models. None of the parameters in any of the population models were found to co-vary with either mesna treatment or metastasised disease.
Correlations between the PK parameters across drugs were evaluated. As elimination rate measurement, CL at the concentration 5 mg/L (5-FU CL) was used in the case of 5-FU. Statistical significant correlations (p<0.05) were found between EPI CL and 5-FU CL and EPI CL and 4-OHCP CL. However, in absolute terms the correlation was weak with r 2-values of 0.1 and 0.05.
The PK/PD model for haematological toxicity
Group IV
The raw data obtained from group IV, 1,017 observations following 329 dosing occasions from 47 individuals, are shown in Fig. 3a. Data from two individuals were excluded due to missing baseline values, whereas data from another two patients were deleted due suspected error in the data. The drug effect caused by EPI was assumed to be the same as had been found in a previous study [41] whereas an attempt to estimate the drug effects induced by 4-OHCP (S 4-OHCP) and 5-FU (S 5-FU) was made. However, since the data did not support the estimation of both slopes and since it turned out that the effect of CP was superior to that of 5-FU, only S 4-OHCP was kept in the model. Using the diagnosis, metastasised breast cancer or not, as a covariate into the model for group IV was not found to be significant. The final parameter estimates are presented in Table 3. The model predicted WBC-time profile in the typical individual of the population after one dosing occasion is described by the solid line in Fig. 3a.

The observed white blood cell count after treatment at occasions that were not followed by G-CSF (a) and those that were followed by treatment with G-CSF (b)
Group III
A total of 1,022 observations from 35 individuals (Fig. 3b) were obtained in group III. Since they all had received G-CSF, an early and rapid increase in WBC-time profiles could be observed. The model developed based on data from group IV described the WBC-time profile poorly. The six alternatives for the influence of G-CSF all improved the fit significantly when added separately. The decreases in the objective function were: 634 (MTT), 582 (addition of new Prol cells), 454 (amplification in maturation chain), 427 (k prol), 284 (γ) and 151 (k circ). Thus, a change in MTT appeared to be the parameter that alone could best describe the change in WBC-time profile when G-CSF was administered. In contrast, and as anticipated based on physiology, changes in k circ could not as well describe changes in the WBC profiles. Various combinations of effects were investigated and it was found that the best model included changes in MTT, k prol and γ. MTT was found to be decreased to 58.4 h and k prol increased by 169% during the entire period of G-CSF administration, while γ was increased to 0.578 during the post-G-CSF period. The increase in the objective function when each one of these components were omitted from the final model was 840, 1,023 and 71 for MTT, k prol and γ, respectively. In some patients a systematic decrease in WBC with time could be observed. A change in k prol over time, estimated to 0.59% per week, best described this change. An IIV of 53% was observed in this parameter. The increase in the objective function value when this component was omitted was 98. In the final model, one of the residual error components was found to be redundant, and the estimated residual error magnitude was 66.7%, when expressed on the linear WBC scale. The parameter estimates of the final model are given in Table 3 and the goodness-of-fit of the model to the observed data are shown in Fig. 4.

Observed and model predicted WBC-time profiles for four patients in group III. The patients were selected based on the average absolute weighted residual, such that the patient with the worst fit (ID=30) and three patients representing the average fit (ranking 16th, 17th and 18th of 35) are shown. Shown are observations (continuous lines), final model predictions based on population (dotted lines) and individual (broken lines) parameter estimates
Discussion
The variability between individuals in elimination capacity has been shown to be extensive for several drugs in cancer patients [9, 16, 33, 39, 48]. Since many anti-cancer drugs have steep dose-response relationships even small deviations in exposure may be of major importance for the final effect. In one study, TDM of methotrexate was found to be superior to traditional dosing in terms of outcome, which indicates the importance of taking the inter-patient variability into account [10]. Hence, developed population PK models, as the models presented in the present study, that include significant covariates that explains part of IIV, can be useful in the design of dosing strategies, not only for TDM purposes but also for the determination of initial doses.
Dosing strategies in anti-cancer therapy based on BSA in adults has been questioned, founded on the fact that BSA seems to be a poor predictor of the elimination capacity of several anti-cancer drugs, including EPI and 5-FU [18–20, 35, 42, 44]. The present study supports this notion. Neither BSA nor weight was significant covariates for the elimination capacity parameters for 5-FU or EPI. However, for 4-OHCP an improvement was obtained when BSA was included to explain part of variability in the CL/fm parameter. In fact, when that covariate was included in the model, together with ALB, an almost 50% decrease in IIV was seen on the CL/fm parameter. The final covariate model for 4-OHCP predicts a 60% difference in clearance between the smallest and the largest individual in terms of BSA, a difference that may have an impact on both response and side effects.
The decomposition of 4-OHCP has earlier been found to be catalysed by albumin in vitro [26]. If that finding can be extrapolated into the in vivo situation, it may explain the present finding that CL/fm for 4-OHCP covaries with ALB. The final covariate model predicts a 40% lower CL/fm in an individual having an ALB of 25 mg/L compared to an individual having 40 mg/L. Serum albumin explained part of variability in the CL of EPI, with increasing elimination at higher albumin levels. In this case, albumin may act as a marker for hepatic function as EPI is eliminated by liver [37]. For 5-FU, the renal function in terms of CLCR co-varied with V max. The kidneys participate in the elimination of 5-FU, although less than 10% of given dose is excreted unchanged in the urine [7]. Previously it has been reported upon that AST better than BILI or other biological markers for liver function reflects the CL of EPI and therefore it has been proposed that EPI dose may be based on AST [8, 45]. That suggestion is not supported by the results in the present study, where BILI was found to be superior to both AST and ALT in explaining inter- and intra-individual variability in PK of EPI.
All structural PK parameters were in accordance with the previously estimated parameters based on data from 21 individuals [40]. In the present study, where 84 patients were included, a less pronounced IIV were found for all three studied drugs. Nonetheless, the ranges of the individual clearances of the drugs studied are still 2–5 fold, which has to be considered as substantial in the context of possible therapeutic consequences. For 4-OHCP, the previous model predicted an IOV of 27%, compared to 14%, the estimate in the present study. However, the previous results were based on only a few individuals from whom PK information from more than one dosing occasion was available, compared to 40 patients in the present analysis.
Anti-cancer dosing regimens usually consist of several cycles of therapy. Thus, the magnitude of IOV is essential to measure and consider, since it determines to what extent PK parameters from one course is applicable across courses. In order to estimate the benefit in terms of improved chance of reaching a target concentration/exposure by performing TDM, the IOV has to be related to IIV. According to the final basic models, the IOV in clearance is less than IIV for all three studied drugs. This indicates that, if a target concentration/exposure was defined, the use of TDM may be meaningful. However, the significant covariates found explained almost 50% of the IIV in CL/fm for 4-OHCP, suggesting that covariate-based dosing of CP may be an alternative.
Only a small difference was found between the variability terms in CL at 5 mg/L for 5-FU in the final covariate model. However, the IIV presented is under-estimated, since data from three individuals having a CL at 5 mg/L that was only 50% of that found in the remaining population, were excluded in the population analysis. We have no explanation for why those individuals differed. However, one reason could be that they were dihydropyrimidine dehydrogenase (DPD) deficient, which has been reported to cause extreme PK profiles for 5-FU [12, 31]. For EPI, the variability between occasions was estimated as being negligible. Although a relatively low IIV in clearance was estimated for EPI, a twofold range in clearance, found in the studied population, may be important in the treatment of the individual patient.
No significant correlation between the elimination rates was found between any of the component drugs in the previous study. The correlations between the clearances for EPI and 4-OHCP and EPI and 5-FU parameters were found to be statistically significant in the present study. However the correlations are still too weak to be considered clinically significant. Hence, monitoring of all three components drugs is necessary if the contribution of each drug to the toxicity or response is to be determined.
A model that describes the relationship between the PK of a drug and the dose-limiting toxicities can be useful, especially in anti-cancer therapy where treatment often has to be aggressive in order to increase the chance of achieving tumour response, and where the patient therefore often experiences severe toxicity. The commonly used approach to study relationships between exposure of anti-cancer drugs and haematological toxicity is to relate the exposure in terms of dose or, in order to account for the variability in elimination capacity, the area under the concentration-time curve (AUC) to the lowest observed white blood cell count that follows a treatment occasion. Relationships between AUC and fractional decrease in leukocyte count at nadir have been derived for drugs administered as single therapy to patients [11, 17, 23]. However, in the polytherapy situation, when more than one of the component drugs are causing bone marrow toxicity, the relationships are more difficult to estimate. One approach is to assume that each drug at standard exposure contributes equally to the toxicity, which was done for the cyclophosphamide-methotrexate-5-FU (CMF) regimen in breast cancer patients [32]. More successful studies have, due to expanded study design possibilities, been performed in animals [5, 43]. In addition, we have recently, using a semi-physiological model, managed to estimate the respective contribution of the EPI and docetaxel to the haematological toxicity in the situation of combination therapy [41].
The haematological toxicity after administration of 5-FU as single agent is moderate in the doses administered within the FEC regimen, although it has been shown to be stronger when given as IV bolus than as constant infusion [21, 27, 29], whereas EPI and CP show strong haematological toxicity on white blood cells. That may be the reason that no significant contribution from 5-FU could be estimated in the present study. This hypothesis is supported by a previous study of the haematological toxicity after administration of the CMF regimen on tumour-bearing rats, where it was found that the haematological effect caused by 5-FU was negligible. The drugs were in that study administered both as single drugs and in all possible combinations, in doses that resulted in concentrations comparable to those achieved in the clinical situation [5].
The PK/PD model describes the data from patients off G-CSF treatment well. Since no single drug data was available for any of the studied drugs, the slope estimate of the haematological toxicity caused by EPI, derived in a previous study on data from metastasised patients treated with EPI and docetaxel, was used in the analysis [41]. This parameter was shown to be able to predict previously presented data [23]. The relative contributions of CP and EPI are in the model estimated under some assumptions. First, linear relationships between drug concentrations and haematological effect are anticipated for both drugs. Although that might not be the case, the data does not support more complex models. Second, the drugs are assumed to act additively under the model. This was found to be an appropriate assumption for the EPI-docetaxel combination and in addition, no known resistance mechanism, as existing for cancer cells, is known for the bone marrow. The estimates of WBCbase (5.7×109/L), MTT (136 h) and γ (0.16) are similar to previous findings, where across six anti-cancer agents the average values were 7×109/L, 125 h and 0.17, respectively [15]. In this paper, we have also quantified the impact of G-CSF on the haematological profile. It has been previously reported that MTT is shortened by G-CSF and in the final model this decrease could be quantified such that MTT is 58 h during G-CSF, compared to 136 h in the absence of G-CSF treatment. Also, G-CSF is known to increase mitotic activity, which in the model was estimated to an increase to 269% of normal activity. Thus, the modelling could from a set of alternative hypotheses, identify realistic mechanisms for the effect during G-CSF treatment and quantify the influence. The increase in the γ parameter in the post-G-CSF period may reflect a true change in the homeostatic capacity of the bone marrow, but could also be a reflection of the fact that without G-CSF, as high leucocyte levels are rarely seen and the system’s response in this range have not previously been determined. It is known that repeated treatment with high-dose chemotherapy and G-CSF can lead to an exhaustion of the bone marrow. In the present model this is described as a continuous decrease, of about 0.6% per week, in Prol activity. This would in the typical patient result in a 10% decrease in the baseline WBC over six courses of chemotherapy.
In summary, we have developed population PK models for all three components drugs of the FEC regimen. In addition, semi-mechanistic PK–PD models, that showed good performance in describing the haematological toxicity after FEC therapy, either alone or in combination with G-CSF, were constructed and resulted in physiologically realistic parameter estimates.
References
Beal S, Sheiner L (1999) NONMEM users guides. University of California, San Fransicso, CA
Bergh J, Wiklund T, Erikstein B, Fornander T, Bengtsson NO, Malmstrom P, Kellokumpu-Lehtinen P, Anker G, Bennmarker H, Wilking N (1998) Dosage of adjuvant G-CSF (filgrastim)-supported FEC polychemotherapy based on equivalent haematological toxicity in high-risk breast cancer patients. Scandinavian Breast Group, Study SBG 9401. Ann Oncol 9:403
Bergh J, Wiklund T, Erikstein B, Lidbrink E, Lindman H, Malmstrom P, Kellokumpu-Lehtinen P, Bengtsson NO, Soderlund G, Anker G, Wist E, Ottosson S, Salminen E, Ljungman P, Holte H, Nilsson J, Blomqvist C, Wilking N (2000) Tailored fluorouracil, epirubicin, and cyclophosphamide compared with marrow-supported high-dose chemotherapy as adjuvant treatment for high-risk breast cancer: a randomised trial. Scandinavian Breast Group 9401 study. Lancet 356:1384
Cockroft D, Gault M (1976) Prediction of creatinine clearance from serum creatinine. Nephron 16:31
De Bruijn EA, Driessen OM, Hermans J (1991) The CMF-regimen. Toxicity patterns following stepwise combinations of cyclophosphamide, methotrexate and fluorouracil. Int J Cancer 48:67
Del Mastro L, Venturini M, Lionetto R, Carnino F, Guarneri D, Gallo L, Contu A, Pronzato P, Vesentini L, Bergaglio M, Comis S, Rosso R (2001) Accelerated-intensified cyclophosphamide, epirubicin, and fluorouracil (CEF) compared with standard CEF in metastatic breast cancer patients: results of a multicenter, randomized phase III study of the Italian gruppo oncologico nord-ouest-mammella inter gruppo group. J Clin Oncol 19:2213
Diasio RB, Harris BE (1989) Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet 16:215
Dobbs NA, Twelves CJ, Gregory W, Cruickshanka C, Richards MA, Rubens RD (2003) Epirubicin in patients with liver dysfunction: development and evaluation of a novel dose modification scheme. Eur J Cancer 39:580
Evans WE, Relling MV (1989) Clinical pharmacokinetics-pharmacodynamics of anticancer drugs. Clin Pharmacokinet 16:327
Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui CH (1998) Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med 338:499
Extra JM, Rousseau F, Bruno R, Clavel M, Le Bail N, Marty M (1993) Phase I and pharmacokinetic study of Taxotere (RP 56976; NSC 628503) given as a short intravenous infusion. Cancer Res 53:1037
Fleming RA, Milano G, Thyss A, Etienne MC, Renee N, Schneider M, Demard F (1992) Correlation between dihydropyrimidine dehydrogenase activity in peripheral mononuclear cells and systemic clearance of fluorouracil in cancer patients. Cancer Res 52:2899
Fossati R, Confalonieri C, Torri V, Ghislandi E, Penna A, Pistotti V, Tinazzi A, Liberati A (1998) Cytotoxic and hormonal treatment for metastatic breast cancer: a systematic review of published randomized trials involving 31,510 women. J Clin Oncol 16:3439
Freyer G, Tranchand B, Ligneau B, Ardiet C, Souquet PJ, Court-Fortune I, Riou R, Rebattu P, Boissel JP, Trillet-Lenoir V, Girard P (2000) Population pharmacokinetics of doxorubicin, etoposide and ifosfamide in small cell lung cancer patients: results of a multicentre study. Br J Clin Pharmacol 50:315
Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO (2002) Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol 15, 20(24):4713
Gamelin E, Boisdron-Celle M, Guerin-Meyer V, Delva R, Lortholary A, Genevieve F, Larra F, Ifrah N, Robert J (1999) Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5-FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: A potential interest for predicting 5-FU toxicity and determining optimal 5-FU dosage. J Clin Oncol 17:1105
Grochow LB, Noe DA, Dole GB, Rowinsky EK, Ettinger DS, Graham ML, McGuire WP, Donehower RC (1989) Phase I trial of trimetrexate glucuronate on a five-day bolus schedule: clinical pharmacology and pharmacodynamics. J Natl Cancer Inst 81:124
Grochow LB, Baraldi C, Noe D (1990) Is dose normalization to weight or body surface area useful in adults? J Natl Cancer Inst 82:323
Gurney H (1996) Dose calculation of anticancer drugs: a review of the current practice and introduction of an alternative. J Clin Oncol 14:2590
Gurney HP, Ackland S, Gebski V, Farrell G (1998) Factors affecting epirubicin pharmacokinetics and toxicity: evidence against using body-surface area for dose calculation. J Clin Oncol 16:2299
Hansen RM, Ryan L, Anderson T, Krzywda B, Quebbeman E, Benson A III, Haller DG, Tormey DC (1996) Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst 88:668
Henderson IC, Hayes DF, Gelman R (1988) Dose-response in the treatment of breast cancer: a critical review. J Clin Oncol 6:1501
Jakobsen P, Bastholt L, Dalmark M, Pfeiffer P, Petersen D, Gjedde SB, Sandberg E, Rose C, Nielsen OS, Mouridsen HT (1991) A randomized study of epirubicin at four different dose levels in advanced breast cancer. Feasibility of myelotoxicity prediction through single blood-sample measurement. Cancer Chemother Pharmacol 28:465
Jonsson EN, Karlsson MO (1998) Automated covariate model building within NONMEM. Pharm Res 15:1463
Karlsson MO, Sheiner LB (1993) The importance of modeling interoccasion variability in population pharmacokinetic analyses. J Pharmacokinet Biopharm 21:735
Kwon CH, Maddison K, LoCastro L, Borch RF (1987) Accelerated decomposition of 4-hydroxycyclophosphamide by human serum albumin. Cancer Res 47:1505
Leichman CG, Fleming TR, Muggia FM, Tangen CM, Ardalan B, Doroshow JH, Meyers FJ, Holcombe RF, Weiss GR, Mangalik A et al (1995) Phase II study of fluorouracil and its modulation in advanced colorectal cancer: a Southwest Oncology Group study. J Clin Oncol 13:1303
Levine MN, Bramwell VH, Pritchard KI, Norris BD, Shepherd LE, Abu-Zahra H, Findlay B, Warr D, Bowman D, Myles J, Arnold A, Vandenberg T, MacKenzie R, Robert J, Ottaway J, Burnell M, Williams CK, Tu D (1998) Randomized trial of intensive cyclophosphamide, epirubicin, and fluorouracil chemotherapy compared with cyclophosphamide, methotrexate, and fluorouracil in premenopausal women with node-positive breast cancer. National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 16:2651
Lokich JJ, Ahlgren JD, Gullo JJ, Philips JA, Fryer JG (1989) A prospective randomized comparison of continuous infusion fluorouracil with a conventional bolus schedule in metastatic colorectal carcinoma: a mid-Atlantic oncology program study. J Clin Oncol 7:425
Microsoft Excel 2000 (1999) Microsoft Corporation
Milano G, Etienne MC (1994) Potential importance of dihydropyrimidine dehydrogenase (DPD) in cancer chemotherapy. Pharmacogenetics 4:301
Moore MJ, Theissen JJ (1992) Cytotoxics and irreversible effects. In: van Boxtel CJ, Holford NHG, Danhof M (eds) The in vivo study of drug action. Elsevier Science Publisher B. V., Amsterdam, p 377
Moore MJ, Erlichman C, Thiessen JJ, Bunting PS, Hardy R, Kerr I, Soldin S (1994) Variability in the pharmacokinetics of cyclophosphamide, methotrexate and 5-fluorouracil in women receiving adjuvant treatment for breast cancer. Cancer Chemother Pharmacol 33:472
Plosker GL, Faulds D (1993) Epirubicin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cancer chemotherapy. Drugs 45:788
Ralph LD, Thomson AH, Dobbs NA, Twelves C (2003) A population model of epirubicin pharmacokinetics and application to dosage guidelines. Cancer Chemother Pharmacol 52:34
Riccardi A, Giordano M, Brugnatelli S, Ucci G, Danova M, Mora O, Fava S, Ascari E (1995) Different doses of epirubicin associated with fixed doses of cyclophosphamide and 5-fluorouracil: a randomised study in advanced breast cancer (letter). Eur J Cancer 31A:1549
Robert J (1994) Clinical pharmacokinetics of epirubicin. Clin Pharmacokinet 26:428
Rodman JH, Abromowitch M, Sinkule JA, Hayes FA, Rivera GK, Evans WE (1987) Clinical pharmacodynamics of continuous infusion teniposide: systemic exposure as a determinant of response in a phase I trial. J Clin Oncol 5:1007
Rodman JH, Relling MV, Stewart CF, Synold TW, McLeod H, Kearns C, Stute N, Crom WR, Evans WE (1993) Clinical pharmacokinetics and pharmacodynamics of anticancer drugs in children. Semin Oncol 20:18
Sandstrom M, Freijs A, Larsson R, Nygren P, Fjallskog ML, Bergh J, Karlsson MO (1996) Lack of relationship between systemic exposure for the component drug of the fluorouracil, epirubicin, and 4-hydroxycyclophosphamide regimen in breast cancer patients. J Clin Oncol 14:1581
Sandstrom M, Lindman H, Nygren P, Lidbrink E, Bergh J, Karlsson MO (2005) Haematological toxicity of the epirubicin-docetaxel regimen in breast cancer patients. J Clin Onc 23:413
Sawyer M, Ratain MJ (2001) Body surface area as a determinant of pharmacokinetics and drug dosing. Invest New Drugs 19:171
Simonsen LE, Wahlby U, Sandstrom M, Freijs A, Karlsson MO (2000) Haematological toxicity following different dosing schedules of 5-fluorouracil and epirubicin in rats. Anticancer Res 20:1519
Terret C, Erdociain E, Guimbaud R, Boisdron-Celle M, McLeod HL, Fety-Deporte R, Lafont T, Gamelin E, Bugat R, Canal P, Chatelut E (2000) Dose and time dependencies of 5-fluorouracil pharmacokinetics. Clin Pharmacol Ther 68:270
Twelves CJ, Dobbs NA, Michael Y, Summers LA, Gregory W, Harper PG, Rubens RD, Richards MA (1992) Clinical pharmacokinetics of epirubicin: the importance of liver biochemistry tests. Br J Cancer 66:765
Van der Wall E, Rutgers EJ, Holtkamp MJ, Baars JW, Schornagel JH, Peterse JL, Beijnen JH, Rodenhuis S (1996) Efficacy of up-front 5-fluorouracil-epidoxorubicin-cyclophosphamide (FEC) chemotherapy with an increased dose of epidoxorubicin in high-risk breast cancer patients. Br J Cancer 73:1080
Venturini M, Del Mastro L, Testore F, Danova M, Garrone O, Lanfranco C, Latini F, Sertoli MR, Lionetto R, Queirolo P, Ardizzoni A, Rosso R (1996) Erythropoietin and granulocyte-macrophage colony-stimulating factor allow acceleration and dose escalation of cyclophosphamide/epidoxorubicin/5-fluorouracil chemotherapy: a dose-finding study in patients with advanced breast cancer. Cancer Chemother Pharmacol 38:487
Yule SM, Boddy AV, Cole M, Price L, Wyllie R, Tasso MJ, Pearson AD, Idle JR (1995) Cyclophosphamide metabolism in children. Cancer Res 55:803
Acknowledgements
First of all we want to thank the patients for participating. We also want to thank Britt Jansson, Malin Johansson, Therese Bäckman, Josefin Ahnström, Malin Lundström, Rita Grönberg, Tina Fornbrandt, Carina Andersson and Leif Astner for assistance in different parts and phases of the work, and the Swedish Cancer Society for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sandström, M., Lindman, H., Nygren, P. et al. Population analysis of the pharmacokinetics and the haematological toxicity of the fluorouracil-epirubicin-cyclophosphamide regimen in breast cancer patients. Cancer Chemother Pharmacol 58, 143–156 (2006). https://doi.org/10.1007/s00280-005-0140-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-005-0140-2