
Abstract
Primary immune thrombocytopenia (ITP) is an intriguing autoimmune disease characterized by autoantibodies against platelets and megakaryocytes. Clinical outcomes, response to treatment, and chronicity predictors were investigated. Patients with newly diagnosed primary ITP treated at a hematology referral center from 2008 to 2018 with complete medical and recent medication history were stratified by age as children < 16 years and adults > 16 years. Responses to treatment including steroids, splenectomy, rituximab, and eltrombopag were classified as response (R) and complete (CR). Factors for developing chronic ITP were determined by multiple regression with uni- and multivariate analysis. p < 0.05 was considered significant. A total of 175 patients were included, 52 children and 123 adults; women predominated with 57.7%. Response to first-line treatment in the whole cohort was 86.18%, CR 43.42% and R 42.76%. The initial response to steroids alone was 83.9% (n = 52/62), rituximab plus high-dose dexamethasone (HDD) 87.2% (n = 34/39), eltrombopag plus HDD 90.9% (n = 10/11), and children receiving IVIG alone 100% (n = 8/8); 9 children were under clinical observation and achieved spontaneous response; loss of response was documented in 15.21% children and 28.3% adults with a median time of 15.95 and 4.07 months respectively; 37.39% of adults and 30.76% of children progressed to a chronic course. Platelets ≥ 20 × 109/L and age ≥ 6 years were risk factors for chronic ITP in the univariate analysis in the adult and children groups, respectively. Clinical course and treatment outcomes for ITP are considerably heterogeneous. Higher platelet counts at diagnosis in adults and age ≥ 6 years in children were associated with an increased risk of chronicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary immune thrombocytopenia (ITP) is a humoral autoimmune condition characterized by the development of autoantibodies against platelet antigens and counts < 100 × 109/L. Its worldwide incidence is 3.3/100,000 person-years [1], with two-thirds of adults and 20 to 25% of children likely to develop a chronic form of the disease [2, 3]. The International Working Group (IWG) classifies ITP as secondary when an underlying disease can be identified, and primary if no such a disease is found [2]. Diagnosis of ITP is based principally on the exclusion of other causes of isolated thrombocytopenia and requires a complete physical examination focusing on bleeding signs with a laboratory workup including a complete blood count (CBC), and hepatitis B and C and human immunodeficiency virus (HIV) testing [4, 5]
The initial event(s) leading to anti-platelet autoimmunity remains unclear, but strong evidence exists that autoantibodies and autoreactive CD8 cytotoxic T cells trigger platelet destruction and also impair platelet production by megakaryocytes in the bone marrow [6]. Autoantibodies directed against platelet glycoproteins have long been accepted as a major pathophysiologic mechanism in ITP, reported for the first time in the classic studies showing the ability of ITP patients’ plasma to cause thrombocytopenia in healthy subjects [7]. However, autoantibodies can also inhibit megakaryocyte proliferation and differentiation, resulting in diminished platelet production. This second mechanism has recently been proposed as an explanation for chronicity [8].
Therapeutic options for newly diagnosed patients consist of treatment for acute bleeding (corticosteroids, intravenous immunoglobulin (IVIG), or anti-D immunoglobulin), causative therapies for immune dysregulation (steroids, rituximab, splenectomy, or Helicobacter pylori eradication), non-causative therapies (thrombopoietin receptor agonists (TPO-RA), danazol), or second-line immunosuppressive therapy (azathioprine, cyclosporine, cyclophosphamide, bortezomib) [5, 9]. Nevertheless, treatment initiation for ITP is not imperative and multiple factors should be considered. This is especially known for children with mild to moderate bleeding who can be managed expectantly with supportive advice and a 24-h contact point, irrespective of platelet count [5].
We report responses to therapy, clinical outcomes, and predictors of chronicity for children and adult patients diagnosed with primary ITP over a decade in an open population from northeast Mexico attending a hematology referral center at a large tertiary care hospital.
Material and methods
There were a total of 258 consecutive patients diagnosed with ITP during the years 2008–2018; in 83 (32%), an underlying disease associated with secondary ITP was found, thus they were not included in the present report. This retrospective study included the remaining 175 patients diagnosed with primary ITP who had complete medical history and electronic files and were treated at the Department of Hematology of the Dr. Jose Eleuterio Gonzalez University Hospital and School of Medicine of the Universidad Autónoma de Nuevo León in Monterrey, Mexico. This is a public institution caring for uninsured patients from the open population. The study was approved by the Institutional Ethics and Research Committee.
Patients were divided into children < 16 years and adults > 16 years with primary ITP in whom a subjacent disease was ruled out. The diagnosis was made according to clinical signs of bleeding and a platelet count < 100 × 109/L [5].
The classification of disease stages was made according to standard terminology including newly diagnosed ITP when the time between clinical symptoms and diagnosis was < 3 months, persistent ITP from 3 to 12 months since diagnosis, and chronic ITP was assumed when the disease lasted ≥ 12 months [2]. A diagnosis of severe ITP required the presence of clinically important bleeding manifestations.
Initial treatment was selected by the treating physician according to patients’ clinical history, comorbidities, bleeding severity, and eligibility for clinical trials.
Treatment protocol options included high-dose dexamethasone (HDD) (40 mg/day for four consecutive doses), weekly intravenous rituximab (100 mg for 4 weeks), and eltrombopag by mouth (50 mg/day for 28 days) [10,11,12,13]. IVIG (1 g/kg on 1 or 2 consecutive days) and danazol (up to 15 mg/kg/day) were alternative therapies administered in children [9]. Prednisone was used alone or combined with doses up to 2.5 mg/kg/day with a maximum dose of 100 mg/day, and then tapered when a rise in the platelet count was obtained along with clinical remission [5, 9].
Responses were classified as complete (CR) when platelets were > 100 × 109/L without bleeding manifestations and response (R) if there was a platelet count > 30 × 109/L or a 2-fold increment from diagnosis without bleeding. Loss of response was defined as a platelet count < 100 × 109/L or less than 2-fold of the baseline platelet count. Refractory ITP was assumed if the patient lost the response after splenectomy [2].
Statistical analysis
The statistical analysis was carried out with SPSS v.22 (IBM SPSS Statistics software, IBM Corp., Armonk, NY). Categorical variables are displayed as absolute numbers and percentages, and comparisons were made with the Pearson x2 test. Quantitative variables were analyzed with descriptive statistics including median and ranges; the Mann–Whitney U test was used for comparisons between quantitative variables. Risk factors for chronicity were studied by logistic regression analysis with a 95% CI. A p < 0.05 was considered significant.
Results
There were 175 patients with primary ITP, 52 (29.7%) children, 123 (70.3%) adults; 101 (57.7%) were women and 74 (42.3%) were men with a median follow-up of 12.8 (1–376) months. Salient characteristics for the whole cohort are shown in Table 1.
Treatment was given to 152 (86.9%) patients, whereas 23 remained under clinical observation due to the low-risk nature of their clinical presentation or idiosyncratic features. Nine children and fourteen adults were part of this non-medical treatment group.
Children
Fifty-two children younger than 16 years with newly diagnosed primary ITP were included. There were 27 (51.9%) girls and 25 (48.1%) boys. Children had a median age of 8 (0–15) years with a median WHO bleeding scale of 1 (0–3). Main clinical characteristics are displayed in Table 1.
Of the 52 children, 21 (40.4%) were treated exclusively with steroids, 8 (15.4%) received IVIG alone, 8 (15.4%) a combination of IVIG plus steroids, 4 (7.7%) rituximab at low doses plus steroids, 2 (3.8%) danazol plus steroids, and 9 (17.3%) children were under clinical observation due to absence of active bleeding. The initial response in this last group was CR in 2 (22.2%) and R in 7 (77.8%).
The initial response in 43 children receiving treatment included 13 (30.2%) CR and 24 (55.8%) R, and 6 (14.0%) were non-responders. The median time to maximum response was 10 (1–18) days. Overall median steroid dose was 1 mg/kg/day (0.25–2.5) with a median time of treatment of 6 (2–24) weeks. Patients received 1.5–2 mg/kg/day for a median time of 3 (2–8) weeks, 1–1.5 mg/kg/day for 4 (2–8) weeks, and < 1 mg/kg/day for 4 (2–8) weeks.
Responses to each treatment and steroid dosage are summarized in Table 2. Three (6.97%) patients had adverse effects secondary to treatment including neutropenia in 2 and Cushing syndrome in one.
Loss of response was documented in 7/46 (15.21%) children; one of them was part of the observational group. Loss of response was found at a median time of 15.95 (0.30–120.4) months with a median number of relapses of 1 (1–2); 4 (57.1%) were treated again with steroids, 2 (28.6%) with rituximab at 100 mg/day for 4 days plus steroids, and danazol in one case (14.3%); all achieved a response.
At the last follow-up, 27 (51.9%) children achieved CR, 13 (25.0%) had persistent disease, and 16 (30.76%) followed a chronic course. The median follow-up for children was 11.0 (range: 1.9–188.0) months.
Risk factors for chronicity were analyzed by logistic regression with a 95% CI. In the univariate analysis, an age ≥ 6 years was statistically significant (shown in Table 3).
Adults
Newly diagnosed ITP was found in 123 adults, 74 (60.2%) women and 49 (39.8%) men. Median age was 42 (16–94) years with a median WHO bleeding scale of 2 (0–3). Clinical characteristics at diagnosis are included in Table 1.
Of the 123 adults, 41 (33.3%) were treated exclusively with steroids, 35 (28.4%) received rituximab at low doses plus steroids, 17 (13.8%) danazol plus steroids, 11 (8.9%) eltrombopag plus HDD, 5 (4.9%) eltrombopag plus HDD plus low doses of rituximab, and 14 (11.3%) were under clinical observation.
Responses documented in 109 patients receiving treatment were 53 (48.6%) CR and 41 (37.6%) R; there were 15 (13.8%) non-responders. The median time to maximum response was 7 (1–45) days. Out of fourteen (11.3%) adults under clinical observation, 4 (28.57%) achieved spontaneous CR, 8 (57.14%) R, and 2 (14.28%) were non-responders. Median cumulative dose of prednisone administered alone was 2485 mg (350–4900) for a median time of 10 (3–24) weeks, whereas median total dosage of prednisone administered with danazol was 2380 mg (140–7280) for a median period of 9 (2–28) weeks. Treatment responses and cumulative doses/week of steroids are summarized in Table 4.
Adverse effects of treatment were observed in 15 (13.76%) adults, mostly associated with steroids, including hyperglycemia in 4, Cushing syndrome in 3, persistent headache in 2, nausea in 2, and weight gain in 2, whereas acne and hirsutism developed in 1 each.
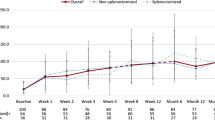
Loss of response was documented in 30/106 (28.3%) patients; one of them belonged to the clinical observation group. The median time to loss of response was 4.07 (0.89–124) months, with a median number of relapses of 1 (1–2); 21 (70%) were treated again with steroids, 7 (23.3%) with rituximab plus HDD, and 2 (6.6%) had a splenectomy. Of the thirty who relapsed, 14 (46.6%) retreated with steroids achieved a second response, whereas one patient was lost to follow-up. Patients treated with rituximab plus HDD (n = 7) and those splenectomized (n = 2) all achieved a response.
At the last follow-up, 58 (41.15%) adults achieved CR, 22 (17.88%) had persistent disease, and 46 (37.39%) progressed to a chronic course. The median follow-up for adults was 12.41 (range: 1.2–376.4) months
Predictors for chronicity were analyzed by logistic regression with a 95% CI. In the univariate analysis, a platelet count ≥ 20 × 109/L was statistically significant, as shown in Table 5.
Discussion
ITP is a common disease in northeast Mexico, being the second and third most frequent hematological diagnosis in children and adults, respectively [14]. Studies dealing with demographic characteristics, clinical findings, treatment alternatives, and long-term clinical outcomes of primary ITP patients in Latin American populations are scarce. Findings in our study cohort contribute to define these features at all ages and help to complete knowledge regarding the heterogeneity of the disease in this region. We gathered information on 175 patients of all ages over a decade.
The treatment landscape for ITP has changed considerably over time in our center. Initial treatment was required for 86.9% patients; this matches the 87.4% demonstrated in a French cohort with a similar sample size [2]. At the last follow-up, 48.5% of patients achieved complete remission regardless of the treatment, close to 49.5% in the French cohort [2]. This confirms a similar pathophysiology for ITP and clinical course across populations.
Steroids alone were favored as first-line treatment in 35.4% of patients with responses of 85.7% in children and 82.9% in adults, close to the reported 85.7% in children [15] and the 80–91% in adults [16, 17]. IVIG alone or with steroids as first-line treatment was highly effective in 87.5% of children, similar to 90% in other studies [18, 19]. Danazol plus steroids was administered in 19 patients with a response of 80%, higher than 51–67% in other reports [20]. Since 2015, when the rituximab era started in ITP, we changed the therapeutic scheme to HDD plus rituximab at low doses with an overall response of 87% [21]. TPO-RAs such as romiplostim and eltrombopag are new targeted therapies recently studied. We have successfully administered eltrombopag plus HDD as initial therapy with a response of 90.7%, higher than the expected > 60% with eltrombopag alone [5]. Eltrombopag plus HDD was previously used with and without rituximab at low doses in adult patients with newly diagnosed ITP [10,11,12,13].
Interestingly, the median time to achieve a response was higher in children than in adults; nevertheless, the response lasted longer in the first group, as observed in the statistically different median time to loss of response of 15 months in children compared with 4 months in adults. Delayed time of response has been speculated to be due to the need of more profound autoantibody reduction and/or to the fact that the main pathogenic action of those autoantibodies consists of impairing platelet production by megakaryocytes in the bone marrow, so megakaryocyte recovery would need to take place first before the platelet count increases [22]. Children in our study also presented lower median platelet counts at diagnosis, 8.6 (range 1.0–76 × 109/L), than adults, 14 (range 0.213–155 × 109/L), as seen in previous studies [23]; however, this difference did not reach significance (p = 0.058).
The later but more enduring response in children could be associated with a lower platelet count at diagnosis predicting a sustained remission [3] but slower recovery to platelet standards. In addition, studies confirm that most children do not have serious bleeding problems, regardless of a lower platelet count, permitting observation as an alternative approach [5]. We did not observe a significant difference in the platelet count at diagnosis between children classified in bleeding stages 1, 2, or 3 (p = 0.145). These findings may indicate that children have a higher platelet threshold for bleeding, perhaps needing a lower limit for the children’s definition of response to treatment, which could also diminish the time to response. Alternative theories involve enhanced platelet function as an explanation to lower bleeding tendency despite lower platelet counts [24]. An example of this is the increased platelet microparticles proposed as a protective mechanism for bleeding in ITP patients, given their role in clot formation [25]. However, further studies are needed to explain children’s better tolerance to thrombocytopenia and their longer response to treatment.
It is of clinical significance if the course of the disease could be predicted at the time of diagnosis. Unfortunately, there are no biological markers that may indicate treatment response, and predictors of chronic ITP have been mainly evaluated in children, with scarce information regarding adults. In our study, the univariate analysis showed age ≥ 6 years as a significant chronicity predictor in children with an HR of 4.333 (95% CI 1.052–17.842). This information relates to a recent meta-analysis of chronicity risk factors in children including 16 studies; patients who developed chronic ITP were older, and the mean difference was 2.68 (95% CI 1.89–3.47) years; age over 8 years was studied with an overall OR for chronic ITP of 2.97 (95% CI 1.42–6.21) [3]. This meta-analysis also demonstrated a higher platelet count and a higher median platelet volume (MPV) as risk factors for chronicity [3]. However, in our study, the platelet count was only accountable as a predictor in the adult group and MPV could not be properly assessed due to abnormally large platelets that outranged blood counter parameters. Larger platelet size appears to be a compensating mechanism of impaired function, as speculated in previous reports [24].
In adult patients, the univariate analysis showed that a platelet count ≥ 20 × 109/L was associated with a higher risk of chronicity with an HR of 5.064 (95% CI 1.315–7.048). In this respect, only one other study took this variable into account and found that platelet counts at diagnosis were significantly higher in the group that developed chronic ITP than in the recovery group, with a 34% increase in odds of chronicity for every 10 × 109/L increase in platelet number [2]. The paradox of a higher platelet count as a risk factor for developing chronic ITP could be related to an epitope-spreading mechanism [26], which is a neoautoreactivity against other epitopes in the original antigenic molecule or to new antigens from inflammatory tissue destruction causing an exacerbation and chronification of autoimmune disease [27]. This phenomenon has been previously associated with the progression of other autoimmune conditions such as systemic lupus erythematosus and type 1 diabetes [28]. The first study describing this mechanism in ITP was recently published and defined epitope spreading as an increase in the number of positive autoantibodies over time. It was observed in 35% of patients with ongoing active disease as opposed to its complete absence in all patients achieving remission. It was also revealed that more glycoproteins targeted predicted higher disease severity [29], suggesting that an upregulation mechanism could be related to chronic ITP; thus, with higher platelet counts, an elevated number of antigenic proteins are present, leading to an increased number of autoantibodies and cytotoxic T lymphocyte production. This could also be triggered by larger platelets in ITP patients, especially in chronic ITP; more platelet surface with more exposed antigens can enhance the immune response. Also, a higher platelet count could reflect a more protracted preclinical course, giving the macrophage and other platelet clearance mechanisms time to refine phagocytic and lytic activities, mediated in part by induction of a higher number of Fc receptors in cells of the phagocytic mononuclear system. Immune characteristics of autoreactive IgG anti-platelet antibodies could be strongly influenced by the type of epitope targeted, as research suggests that different types of antibodies may differentially alter clearance, inhibit megakaryopoiesis, or induce platelet apoptosis [30]. Research of epitope spreading as a self-perpetuating mechanism in ITP could have the potential for developing targeted therapies to prevent and treat chronic disease.
Another possible mechanism for a higher platelet count as a risk factor for chronicity is reduced thrombopoietin (TPO) reaching the bone marrow. This is explained by a low platelet count promoting TPO production in the liver. However, circulating TPO binds to platelets via the Mpl receptor promoting its rapid clearance [6]. Therefore, more platelets in the bloodstream could prevent already reduced TPO from reaching the bone marrow to stimulate new platelet production, perpetuating thrombocytopenia.
A new hypothesis on chronicity involves exacerbated platelet apoptosis and increased loss of sialic acid [31, 32]. This acid is normally lost in age terminal platelets to expose underlying galactose residues allowing clearance by the Ashwell–Morrel receptor (AMR) in hepatocytes [6].
Principal limitations in this study are its retrospective nature and a relatively limited number of patients. The main strengths are an adequate length of follow-up, stringent documentation, and a robust statistical analysis. In addition, studying platelet laboratory findings in thrombocytopenia has been known to be challenging. In this case, MPV could not be properly measured in all patients due to the presence of large platelets that exceeded the laboratory parameters set for the blood automated counter.
Conclusion
ITP is a highly complex disease; its pathophysiology and initial events are intricate and not fully understood, with multiple theories being brought up to explain a chronic course. Response to therapy remains heterogeneous and unpredictable; nevertheless, outcomes have improved over time. Further research is needed to confirm predictive factors for chronicity so treatment and follow-up could be refined, improving the quality of life of ITP patients.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Frederiksen H, Christiansen CF, Norgaard M (2012) Risk and prognosis of adult primary immune thrombocytopenia. Expert Rev Hematol 5:219–228. https://doi.org/10.1586/ehm.12.7
Grimaldi-Bensouda L, Nordon C, Michel M, Viallard JF, Adoue D, Magy-Bertrand N, Durand JM, Quittet P, Fain O, Bonnotte B, Morin AS, Morel N, Costedoat-Chalumeau N, Pan-Petesch B, Khellaf M, Perlat A, Sacre K, Lefrere F, Abenhaim L, Godeau B, Group for the PGRx-ITP Study (2016) Immune thrombocytopenia in adults: a prospective cohort study of clinical features and predictors of outcome. Haematologica 101:1039–1045. https://doi.org/10.3324/haematol.2016.146373
Heitink-Pollé KMJ, Nijsten J, Boonacker CWB, De Haas M, Bruin MCA (2014) Clinical and laboratory predictors of chronic immune thrombocytopenia in children: a systematic review and meta-analysis. Blood 124:3295–3307. https://doi.org/10.1182/blood-2014-04-570127
Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, Bussel JB, Cines DB, Chong BH, Cooper N, Godeau B, Lechner K, Mazzucconi MG, McMillan R, Sanz MA, Imbach P, Blanchette V, Kühne T, Ruggeri M, George JN (2009) Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 113:2386–2393. https://doi.org/10.1182/blood-2008-07-162503
Provan D, Arnold DM, Bussel JB, Chong BH, Cooper N, Gernsheimer T, Ghanima W, Godeau B, González-López TJ, Grainger J, Hou M, Kruse C, McDonald V, Michel M, Newland AC, Pavord S, Rodeghiero F, Scully M, Tomiyama Y, Wong RS, Zaja F, Kuter DJ (2019) Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv 3:3780–3817. https://doi.org/10.1182/bloodadvances.2019000812
Swinkels M, Rijkers M, Voorberg J, Vidarsson G, Leebeek FWG, Jansen AJG (2018) Emerging concepts in immune thrombocytopenia. Front Immunol 9:880. https://doi.org/10.3389/fimmu.2018.00880
Harrington WJ, Sprague CC, Moore CV, Aulvin RC, Dubach R (1953) Immunologic mechanisms in idiopathic and neonatal thrombocytopenic purpura. Ann Intern Med 38:433–469. https://doi.org/10.1258/acb.2012.201227
Nugent D, McMillan R, Nichol JL, Slichter SJ (2009) Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol 146:585–596. https://doi.org/10.1111/j.1365-2141.2009.07717.x
Neunert C, Terrell DR, Arnold DM, Buchanan G, Cines DB, Cooper N, Cuker A, Despotovic JM, George JN, Grace RF, Kühne T, Kuter DJ, Lim W, McCrae KR, Pruitt B, Shimanek H, Vesely SK (2019) American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv 3:3829–3866. https://doi.org/10.1182/bloodadvances.2019000966
Gómez-Almaguer D, Colunga-Pedraza PR, Gómez-De León A, Gutiérrez-Aguirre CH, Cantú-Rodríguez OG, Jaime-Pérez JC (2019) Eltrombopag, low-dose rituximab, and dexamethasone combination as frontline treatment of newly diagnosed immune thrombocytopaenia. Br J Haematol 184:288–290. https://doi.org/10.1111/bjh.15070
Gómez-Almaguer D, Tarín-Arzaga L, Moreno-Jaime B, Jaime-Pérez JC, Ceballos-López AA, Ruiz-Argüelles GJ, Ruiz-Delgado GJ, Cantú-Rodríguez OG, Gutiérrez-Aguirre CH, Sánchez-Cárdenas M (2013) High response rate to low-dose rituximab plus high-dose dexamethasone as frontline therapy in adult patients with primary immune thrombocytopenia. Eur J Haematol 90:494–500. https://doi.org/10.1111/ejh.12102
Gómez-Almaguer D, Herrera-Rojas MA, Jaime-Pérez JC, Gómez-de León A, Cantú-Rodríguez OG, Gutiérrez-Aguirre CH, Tarín-Arzaga L, Hernández-Reyes J, Ruiz-Arguelles GJ (2014) Eltrombopag and high-dose dexamethasone as frontline treatment of newly diagnosed immune thrombocytopenia in adults. Blood 123:3906–3908. https://doi.org/10.1182/blood-2014-01-549360
Gómez-Almaguer D (2018) Eltrombopag-based combination treatment for immune thrombocytopenia. Ther Adv Hematol 9:309–317. https://doi.org/10.1177/2040620718798798
Jaime-Pérez JC, Treviño-Reyna G, Aguilar-Calderón P, Cantú-Rodríguez OG, Marfil-Rivera LJ, Gómez-Almaguer D (2018) Contributions of a regional approach to document hematologic disease in Mexico: a 10-year experience in an open population. Hematology 23:803–809. https://doi.org/10.1080/10245332.2018.1498166
Mazzucconi MG, Fazi P, Bernasconi S, de Rossi G, Leone G, Gugliotta L, Vianelli N, Avvisati G, Rodeghiero F, Amendola A, Baronci C, Carbone C, Quattrin S, Fioritoni G, D'Alfonso G, Mandelli F, for the Gruppo Italiano Malattie EMatologiche dell'Adulto (GIMEMA) Thrombocytopenia Working Party (2007) Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 109:1401–1407. https://doi.org/10.1182/blood-2005-12-015222
Nomura S (2016) Advances in diagnosis and treatments for immune thrombocytopenia. Clin Med Insights Blood Disord 9:15–22. https://doi.org/10.4137/CMBD.S39643
Chang H, Tang TC, Hung YS, Li PL, Kuo MC, Wu JH, Wang PN (2018) Immune thrombocytopenia: effectiveness of frontline steroids and comparison of azathioprine, splenectomy, and rituximab as second-line treatment. Eur J Haematol 101:549–555. https://doi.org/10.1111/ejh.13144
Bussel JB, Eldor A, Kelton JG, Varon D, Brenner B, Gillis S, Angiolillo A, Kulkarni R, Abshire T, Kelleher J, the IGIV-C in ITP Study Group (2004) IGIV-C, a novel intravenous immunoglobulin: evaluation of safety, efficacy, mechanisms of action, and impact on quality of life. Thromb Haemost 91:771–778. https://doi.org/10.1160/TH03-10-0650
Arnold DM (2013) Positioning new treatments in the management of immune thrombocytopenia. Pediatr Blood Cancer 60:S19–S22. https://doi.org/10.1002/pbc.24341
Zimmer J (2019) Danazol as a second-line treatment for immune thrombocytopenic purpura. Eur J Haematol 102:97–98. https://doi.org/10.1111/ejh.13184
Zaja F, Battista ML, Pirrotta MT, Palmieri S, Montagna M, Vianelli N, Marin L, Cavallin M, Bocchia M, Defina M, Ippoliti M, Ferrara F, Patriarca F, Avanzini MA, Regazzi M, Baccarani M, Isola M, Soldano F, Fanin R (2008) Lower dose rituximab is active in adults patients with idiopathic thrombocytopenic purpura. Haematologica 93:930–933. https://doi.org/10.3324/haematol.12206
Newland AC, Sánchez-González B, Rejtő L, Egyed M, Romanyuk N, Godar M, Verschueren K, Gandini D, Ulrichts P, Beauchamp J, Dreier T, Ward ES, Michel M, Liebman HA, Haard H, Leupin N, Kuter DJ (2020) Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am J Hematol 95:178–187. https://doi.org/10.1002/ajh.25680
Kühne T, Berchtold W, Michaels LA et al (2011) Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the intercontinental cooperative immune thrombocytopenia study group. Haematologica 96:1831–1837. https://doi.org/10.3324/haematol.2011.050799
Nobuko N, Hirokazu K, Akuta K et al (2020) Reevaluation of platelet function in chronic immune thrombocytopenia: impacts of platelet size, platelet-associated anti-αIIbβ3 antibodies and thrombopoietin receptor agonists. Br J Haematol 189:760–771. https://doi.org/10.1111/bjh.16439
Boulware R, Refaai MA (2020) Why do patients with immune thrombocytopenia (ITP) experience lower bleeding events despite thrombocytopenia? Thromb Res 187:154–158. https://doi.org/10.1016/j.thromres.2020.01.020
Cooper N, Bussel J (2006) The pathogenesis of immune thrombocytopaenic purpura. Br J Haematol 133:364–374. https://doi.org/10.1111/j.1365-2141.2006.06024.x
Cines DB, Blanchette VS (2002) Immune thrombocytopenic purpura. N Engl J Med 346:995–1008. https://doi.org/10.1056/NEJMra010501
Venkatesha SH, Durai M, Moudgil KD (2015) Epitope spreading in autoimmune diseases. In: Shoenfeld Y, Agmon-Levin N, Rose NR (eds) Infection and Autoimmunity, 2nd. edn. Academic press, Amsterdam, pp 45–68. https://doi.org/10.1016/B978-0-444-63269-2.00003-9\
Al-Samkari H, Rosovsky RP, Leaf RSK et al (2020) A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv 4:9–18. https://doi.org/10.1182/bloodadvances.2019000868
Lambert MP, Gernsheimer TB (2017) Clinical updates in adult immune thrombocytopenia. Blood 129:2829–2835. https://doi.org/10.1182/blood-2017-03-754119
Monzón Manzano E, Álvarez Román MT, Justo Sanz R, Fernández Bello I, Hernández D, Martín Salces M, Valor L, Rivas Pollmar I, Butta NV, Jiménez Yuste V (2020) Platelet and immune characteristics of immune thrombocytopaenia patients non-responsive to therapy reveal severe immune dysregulation. Br J Haematol 189:943–953. https://doi.org/10.1111/bjh.16459
Li J, Van Der Wal DE, Zhu G et al (2015) Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun 6:7737. https://doi.org/10.1038/ncomms8737
Acknowledgments
We thank Sergio Lozano-Rodriguez, MD, for his critical review of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Lorena Salazar-Cavazos, Patrizia Aguilar-Calderón, and Raúl A. Jiménez-Castillo. The first draft of the manuscript was written by Eugenia M. Ramos-Dávila, David Gómez-Almaguer, and José Carlos Jaime-Pérez. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
The protocol of the study was approved by the Ethics and Research Committee of the institution and is in full compliance with the principles of the Declaration of Helsinki as revised in 2013.
Consent to participate
The Ethics and Research Committee of the institution waved this consent due to its retrospective nature.
Consent for publication
The Ethics and Research Committee of the institution waved this consent due to its retrospective nature and anonymized information.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jaime-Pérez, J.C., Aguilar-Calderón, P., Jiménez-Castillo, R.A. et al. Treatment outcomes and chronicity predictors for primary immune thrombocytopenia: 10-year data from an academic center. Ann Hematol 99, 2513–2520 (2020). https://doi.org/10.1007/s00277-020-04257-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-020-04257-2