
Abstract
Our aim was to evaluate the clinical value of splenectomy as a treatment for relapsed hemophagocytic lymphohistiocytosis (HLH) of unknown cause in adults. We retrospectively reviewed the clinical data from medical records of 19 adults with relapsed HLH of unknown cause treated with splenectomy in our institution from June 2007 to March 2014. To rule out possible underlying diseases, including infection, autoimmune disease, neoplasms, and primary HLH, the patients had undergone examinations including F18 fluoro-2-deoxyglucose positron emission tomography/computed tomography, HLH-associated gene defects, and lymph node biopsies. Twelve patients (63.2 %) achieved partial responses (PR), whereas seven patients (36.8 %) had no response (NR) prior to splenectomy. Infection and hemorrhage were the main complications of splenectomy. Eighteen cases were evaluable after follow-up. Seven cases with histopathologic diagnoses of lymphoma had received chemotherapy, four of whom had achieved complete responses (CR), one PR, and two NR. Maintenance treatment was ceased 2 or 3 months after splenectomy in the other 11 cases, five of whom had CR, four PR, and two NR. Eleven of 18 cases (61.1 %) survived with a median follow-up of 25 months (range 3–79 months) for survivors. Twelve- and 36-month progression-free survival rates were 48 and 24 %, respectively; 12- and 36-month overall survival rates were 57 and 25 %, respectively. Median survival time was 22 months. Our results indicate splenectomy may be an effective means of diagnosis and treatment of relapsed HLH of unknown cause. Further study is required to establish the mechanism and value of splenectomy in this disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a hyperinflammatory syndrome caused by hereditary or acquired immune dysregulation [1]. In this severe immune dysregulation disorder, lymphocytes and histiocytes become overactive and secrete excessive amounts of inflammatory cytokines. HLH manifests clinically as fever, hepatosplenomegaly, and pancytopenia and is classified as either primary HLH or secondary HLH according to the underlying cause. Secondary HLH is a reactive disorder that can be initiated by a large variety of diseases that activate the immune system, such as infection, malignancy, and rheumatic disease. For this reason, all HLH patients should be assessed for such underlying diseases. Even so, some patients are still diagnosed as having HLH of unknown cause. HLH-2004, published by the Histiocyte Society, is the current therapeutic guideline for HLH [2]. In spite of recommended treatment, there are many cases of refractory/relapsed HLH [3]. Thus, diagnosis and treatment remain challenges for adults with relapsed HLH of unknown cause. There is a lack of data concerning valid and effective second-line therapies and criteria for evaluating their therapeutic efficacy [3].
In healthy humans, the spleen is an important part of the lymphatic system and harbors large amounts of major immunocytes and cytokines. It is the largest lymphatic organ and the only lymphoid tissue that acts as a blood filter. The spleen not only functions in specific and nonspecific immunity, produces lymphocytes, and secretes large amount of antibodies but it also removes aged cells and microbial antigens from the blood thus helping to maintain homeostasis [4]. However, the spleen can be affected by hematologic diseases and plays an important role in disease occurrence and development. Hence, splenectomy has an important role in the treatment of hematologic diseases. Given that the spleen contains large amounts of important immunocytes and cytokines and HLH is believed to be caused by a cytokine storm resulting from immune dysregulation, splenectomy would likely control HLH through adjusting inflammation and concentrations of inflammatory factors. Splenectomy as a new salvage therapy for HLH has not been widely reported [5, 6].
Herein, we report a retrospective review and evaluation of the medical records of 19 adults with relapsed HLH of unknown cause who were treated in the Department of Hematology, Beijing Friendship Hospital, from June 2007 to March 2014.
Methods
Patients
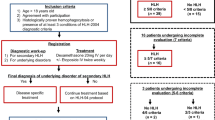
In all, 201 adults with HLH were treated in the Department of Hematology, Beijing Friendship Hospital, between June 2007 and March 2014. The causes had been established in 170 of these patients and included infection, rheumatic and immunologic diseases, malignant tumors (especially lymphoma), and primary HLH. Nineteen of the remaining 31 patients with HLH of unknown origin had been treated with splenectomy and were therefore enrolled in this study. All 19 patients had experienced relapses of HLH after initial remissions achieved with standard HLH therapy and had failed to achieve complete responses with salvage therapies.
All study cases had received one or more types of salvage therapies prior to splenectomy. The therapeutic regimens included (1) high-dose glucocorticosteroid + etoposide +/− cyclosporine A, (2) high-dose glucocorticosteroid + etoposide +/− gamma globulin, (3) FD regimens (fludarabine + glucocorticosteroid), (4) ECHOP-like regimens (etoposide + cyclophosphamide + epirubicin + vincristine + glucocorticosteroid), and ECOP-like regimens (etoposide + cyclophosphamide + vincristine + glucocorticosteroid). The patients were not able to undergo hematopoietic stem cell transplantation for various reasons.
This study was approved by the Ethics Committee of Beijing Friendship Hospital, and because it was a retrospective review of medical records, the requirement for informed patient consent was waived.
Diagnostic criteria
The diagnoses of HLH had been made according to the HLH-2004 diagnostic criteria [2], namely, a molecular diagnosis of HLH or five of the following eight criteria met (1) fever with temperature greater than 38.5 °C for at least 7 days; (2) splenomegaly (≥3 cm below the costal margin); (3) cytopenia not caused by impaired hematopoiesis in the bone marrow and involving at least two of three lineages (hemoglobin <90 g/L, platelet count <100 × 109/L, or neutrophil count <1.0 × 109/L); (4) hypertriglyceridemia and/or hypofibrinogenemia (fasting triglycerides >3 mmol/L or fibrinogen <1.5 g/L); (5) hemophagocytosis detected in the bone marrow, spleen, or lymph nodes; (6) low or absent natural killer cell activity; (7) total serum ferritin concentrations ≥500 μg/L; and (8) high concentrations of soluble interleukin-2 receptor (sCD25).
Investigations
All enrolled patients had undergone medical examinations including F-18 fluoro-2-deoxyglucose positron emission tomography/computed tomography (F18-FDG PET/CT), HLH-associated genes, and lymph node biopsies to exclude infection, rheumatic and immunologic diseases, malignant tumors (especially lymphoma), and primary HLH. The clinical characters and laboratory findings were retrospectively evaluated by reviewing patient medical records.
Assessment of response
Overall survival (OS) was defined as the time from splenectomy to death from any cause. Progression-free survival (PFS) was defined as the time from splenectomy to disease progression or death. A complete response (CR) [3] was defined as absence of clinical symptoms and reversion to normal values of all quantifiable symptoms and laboratory markers of HLH. A partial response (PR) was defined as at least a 25 % improvement in two or more quantifiable symptoms or laboratory markers: sCD25, serum ferritin, or triglyceride concentrations. For patients with an initial absolute neutrophil count less than 0.5 × 109/L, a PR was defined as an absolute neutrophil count fully recovered to greater than 0.5 × 109/L. For patients with ALT greater than 400 U/L, a response was defined as a reduction of ALT by ≥50 %. Resolution of hemophagocytosis was noted in all patients. Consciousness had recovered to normal in patients with refractory central nervous system HLH and altered levels of consciousness. A no response (NR) was defined as failing to achieve PR. Responses were evaluated every 2 weeks postsplenectomy until the study endpoints or death.
Statistical analysis
Statistical analysis was performed with SPSS version 16.0 (SPSS, Chicago, IL, USA). Data are presented as the mean ± standard deviation (SD) or percentage. Survival was estimated by the Kaplan–Meier method. The log-rank test was used to compare survival times. Values of p < 0.05 were statistically significant.
Results
Clinical features
The preoperative clinical features of the 19 patients with HLH of unknown cause are summarized in Table 1. They comprised 13 men and six women aged between 17 and 63 years (mean age of 35.2 ± 16.2 years). All patients (100 %) presented with fever, abnormal liver function, and cytopenia in two or more lineages. Eighteen patients (94.7 %) presented with splenomegaly, 14 (73.7 %) with coagulation disorder, and 10 (52.6 %) with hemophagocytosis in the bone marrow and/or lymph nodes.
Prior to their splenectomies, 12 of the 19 patients (63.2 %) had experienced PRs whereas the other seven (36.8 %) had had NRs prior to therapies.
F18-FDG PET/CT findings and histopathology of spleen
All 19 patients underwent F18-FDG PET/CT scan to assess their spleens, and 18 were found to have enlarged spleens. The remaining patient’s spleen was of normal size but was found to be congested on histopathologic examination. Increased radioactivity distribution was present in eight patients. Six of those eight patients were diagnosed histopathologically with lymphoma. Radioactivity distribution was not uniformly increased in the three patients who were found on histopathologic examination of the spleen to have diffuse large B-cell lymphoma. No abnormalities in radioactivity distribution were detected in 11 patients, one of whom was diagnosed with T-cell lymphoma on histopathologic examination of the spleen.
Radioactivity distribution in central and periphery bone marrow was diffusely increased (standardized uptake value [SUV] 3.9–5.9) in seven patients in whom the spleen histopathology verified lymphoma. However, in these patients, bone marrow biopsy and flow cytometry provided no evidence of lymphoma. Radioactivity distribution in the liver was increased in two patients (nos. 11 and 15, Table 2). F18-FDG PET/CT revealed no foci of increased SUV outside the spleen in the remaining patients.
The histopathologic findings in the spleens of the 19 patients are summarized in Table 2: seven cases of lymphoma, four of extramedullary hematopoiesis, three of histiocytic proliferation with hemophagocytosis, three of splenic congestion, one of hypersplenism, and one of atypical hyperplasia of T-cells.
Postsplenectomy complications
The major postsplenectomy complications of HLH patients were infection and bleeding. Two patients experienced severe pulmonary infection, one of them recovered after antibiotic treatment and the other died 3 days postoperatively of pulmonary infection with respiratory failure. One patient experienced extensive oozing of blood from the wound. Initially, this patient’s condition did not improve with massive transfusion of platelets, fresh frozen plasma, prothrombin complex, and fibrinogen; however, after supplemental factor VII, this patient eventually recovered.
Outcomes
One of the 19 patients was lost to follow-up; thus, 18 patients were evaluable. Of the seven patients with lymphoma, four patients achieved CR, one PR, and two NR with chemotherapy. The 11 evaluable patients without lymphoma received treatment for 2–3 months following splenectomy: five achieved CR, four achieved PR, and two NR (Table 1). The five patients with CR achieved long-term disease-free survival, the longest so far being in a 17-year-old male patient who has survived for 57 months. Of the four patients with PR, one patient relapsed 18 months after splenectomy and another was found to have subcutaneous panniculitis-like T-cell lymphoma 1 year postsplenectomy.
The median duration of follow-up for survivors was 25 months (range 3–79 months). Eleven patients (61.1 %) survived, and seven patients (38.9 %) died. Five of these patients died of the primary disease and one from pulmonary infection after splenectomy. The seventh experienced a relapse of HLH, underwent nonidentical allogeneic hematopoietic stem cell transplantation, and died of complications of transplantation.
The rates of PFS at 12 and 36 months following splenectomy were 48 and 24 %, respectively (Fig. 1a). The rates of OS at 12 and 36 months following splenectomy were 57 and 25 %, respectively (Fig. 1b). The median survival time following splenectomy was 22 months. There was no significant difference in survival times between patients with and without underlying lymphoma (log-rank test, p = 0.283) (Fig. 2).

PFS (a) and OS (b) of HLH after splenectomy (n = 18)

Survival times between patients with and without underlying lymphoma (log-rank test, p = 0.283)
Discussion
All patients with HLH should be assessed for underlying diseases that may have induced this disease. However, some patients will still be diagnosed as having HLH of unknown cause. We previously retrospectively reviewed 72 patients with HLH treated in our hospital [7]; we did not identify a cause in 6.9 % of those patients. According to the criteria of Das Gupta et al. [8], a diagnosis of primary splenic lymphoma (PSL) can be made when the lymphoma is confined to the spleen or splenic hilar lymph nodes. PSL occurs in less than 1 % of all cases of non-Hodgkin lymphomas [9]. It is even rarer for HLH to occur in combination with PSL. Splenectomy provides the diagnosis and is the main treatment for PSL. Han et al. [10] reported a 77-year-old man who presented with HLH, experienced spontaneous splenic rupture after chemotherapy, and was diagnosed as having PSL upon histopathologic examination of his spleen after emergency splenectomy. Suzuki et al. [11] reported a patient with HLH who was found to have aggressive natural killer cell leukemia after a splenectomy performed after 3 years of immunosuppressive therapy. In the current study, seven of 19 patients with HLH (36.8 %) were diagnosed with lymphoma by histopathologic examination of their spleens. These findings suggest that PSL may be the cause of HLH in some patients, in which case, splenectomy would provide both a diagnosis and treatment. Ongoing chemotherapy would likely prolong their survival times.
F18-FDG PET/CT provides both functional (PET scan) and anatomic (CT imaging) information and thus facilitates diagnosis of this disease. Kim J. et al. [12] reported that the sensitivity and specificity of F18-FDG PET/CT for diagnosis of HLH are 83 and 71 %, respectively. Specifically, enlarged superficial lymph nodes are rarely detected in lymphoma-associated HLH, which often involves extranodal sites, deep tissues, and organs, the spleen being the most commonly involved. All 19 patients in the current study had undergone F18-FDG PET/CT examination, and 18 had been found to have enlarged spleens. The remaining patient had a normal-sized spleen in which congestion was found histopathologically. Increased radioactivity distribution was present in eight patients, six of whom were diagnosed with lymphoma based on histopathologic findings. Radioactivity distribution was not uniformly increased in three patients whose spleens contained diffuse large B-cell lymphoma. No abnormalities in radioactivity distribution were detected in 11 patients, one of whom had a histologic diagnosis of T-cell lymphoma. These findings indicate that F18-FDG PET/CT identification of an enlarged spleen and increased radioactivity distribution, especially when it is nonuniform, strongly suggests splenic lymphoma.
HLH-2004, published by the Histiocyte Society, is the current therapeutic guideline for HLH [2]. Many patients develop refractory/relapsed HLH. There is a lack of valid and effective second-line therapies and criteria for evaluating therapeutic efficacy [3]. Various institutions have reported different salvage therapies, including CHOP-like regimens [13], high-dose glucocorticosteroid combined with etoposide [14], FD regimens [7] with or without gamma globulin, rituximab [15], alemtuzumab [2], and allogeneic hematopoietic stem cell transplantation [16]. All 19 patients in our study had experienced relapses of HLH after initial remissions induced by standard HLH therapy and had failed to achieve CRs after two or more salvage therapies. Only a few individual cases of therapeutic splenectomy as a salvage therapy for HLH have been reported [5, 6]. Imashuku et al. [5] reported five patients with childhood HLH who underwent splenectomy. Zhang et al. [6] present a case diagnosed as sHLH who has recovered from HLH following comprehensive treatment based on splenectomy. Zhang et al. [17] found that the anti-inflammatory effects of splenectomy protected rats from cerebral damage after stroke. After splenectomy, fewer T-cells, neutrophils, and macrophages were observed in brain tissue and serum concentrations of pro-inflammatory cytokines such as interleukin and tumor necrosis factor decreased. Chu et al. [18] demonstrated that splenectomy downregulates the mitogen-activated protein kinase-nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway in rat brains after traumatic brain damage. Chuang et al. [19] reported that Epstein–Barr virus-associated T- or natural killer/T-cell lymphoma shows constitutive activation of NF-κB, explaining its hypercytokinemia and poor prognosis. We therefore postulated that inhibition of the NF-κB signal pathway after splenectomy was a potential target for the treatment of HLH. Twelve of 19 HLH patients in our current study did not have associated lymphoma. Following splenectomy, five of these patients achieved CR and four PR. The five patients with CR achieved long-term disease-free survival, the longest so far being in a 17-year-old male patient who has survived for 57 months. Our results suggest that patients who undergo splenectomy experience high CR rates and that their long-term outcomes are superior. At present, there are few published data regarding second-line therapies and survival after relapse of HLH of unknown cause. Notably, most deaths occur during the first few weeks of treatment and may reflect either preexisting morbidities or primary refractory disease [20]. In our study, 11 patients (61.1 %) survived and seven (38.9 %) died during follow-up (one lost to follow-up). Our results (Fig. 1) suggest that splenectomy is an effective treatment for this disease. The main causes of death were the primary diseases. There were no significant differences in survival times between patients with and without underlying lymphoma (Fig. 2); however, this result is inconclusive because of the small number of patients enrolled in this study.
All patients in this study (100 %) presented with fever, abnormal liver function, and cytopenia in two or more lineages. Coagulation disorders were detected in 14 patients (73.7 %). Compared with patients without HLH, the risk of splenectomy is significantly greater in those with HLH. To reduce this risk, all our patients received salvage therapies in an attempt to achieve PR prior to splenectomy. Infection and bleeding are the major postsplenectomy complications. One patient who had pulmonary infection prior to surgery was found to have diffuse large B-cell lymphoma on pathologic examination of his spleen and died 3 days postsplenectomy of pulmonary infection with respiratory failure. Thus, there was no opportunity to treat this patient’s primary disease. Another patient with preoperative coagulation disorder experienced extensive blood oozing from the wound after splenectomy. These cases suggest that attempts should be made to control infection and correct coagulation disorders prior to splenectomy to reduce the risk of surgery.
In conclusion, splenectomy may be an effective means of diagnosing and treating relapsed adult HLH of unknown cause. Further studies are required to identify the mechanisms of action and the outcomes of performing splenectomy in patients with this disease.
References
Verbsky JW, Grossman WJ (2006) Hemophagocytic lymphohistiocytosis: diagnosis, pathophysiology, treatment, and future perspectives. Ann Med 38:20–31
Henter JI, Horne A, Aricó M et al (2007) HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48:124–131
Marsh RA, Allen CE, McClain KL et al (2013) Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer 60:101–109
Tarantino G, Savastano S, Capone D et al (2011) Spleen: a new role for an old player? World J Gastroenterol 17:3776–3784
Imashuku S, Obayashi M, Hosoi G et al (2000) Splenectomy in haemophagocytic lymphohistiocytosis: report of histopathological changes with CD19+ B-cell depletion and therapeutic results. Br J Haematol 108:505–510
Zhang LJ, Zhang SJ, Xu J (2011) Splenectomy for an adult patient with refractory secondary hemophagocytic lymphohistiocytosis. Biomed Pharmacother 65:432–435
Wang YN, Wang Z, Wu L et al (2009) A multicenter retrospective analysis of diagnosis and treatment of 72 hemophagocytic syndrome patients. Chin J Hematol 30:793–798
Das-Gupta T, Coombes B, Brasfield RD (1965) Primary malignant neoplasms of the spleen. Surg Gynecol Obstet 120:947–960
Brox A, Shustik C (1993) Non-Hodgkin’s lymphoma of the spleen. Leuk Lymphoma 11:165–171
Han SM, Teng CL, Hwang GY et al (2008) Primary splenic lymphoma associated with hemophagocytic lymphohistiocytosis complicated with splenic rupture. J Chin Med Assoc 71:210–213
Suzuki S, Uozumi K, Utsunomiya A et al (2008) Aggressive NK cell leukaemia after splenectomy: association with CD95-resistant memory T-cell proliferation and recalcitrant clinical course of haemophagocytic syndrome. Eur J Haematol 81:236–241
Kim J, Yoo SW, Kang SR et al (2014) Clinical implication of F-18 FDG PET/CT in patients with secondary hemophagocytic lymphohistiocytosis. Ann Hematol 93:661–667
Shin HJ, Chung JS, Lee JJ et al (2008) Treatment outcomes with CHOP chemotherapy in adult patients with hemophagocytic lymphohistiocytosis. J Korean Med Sci 23:439–444
Dunn T, Cho M, Medeiros B et al (2012) Hemophagocytic lymphohistiocytosis in pregnancy: a case report and review of treatment options. Hematology 17:325–328
Chellapandian D, Das R, Zelley K et al (2013) Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol 162:376–382
Adachi S, Kubota M, Akiyama Y et al (1997) Successful bone marrow transplantation from an HLA-identical unrelated donor in a patient with hemophagocytic lymphohistiocytosis. Bone Marrow Transplant 19:183–185
Zhang BJ, Men XJ, Lu ZQ et al (2013) Splenectomy protects experimental rats from cerebral damage after stroke due to anti-inflammatory effects. Chin Med J (Engl) 126:2354–2360
Chu W, Li M, Li F et al (2013) Immediate splenectomy down-regulates the MAPK-NF-kappaB signaling pathway in rat brain after severe traumatic brain injury. J Trauma Acute Care Surg 74:1446–1453
Chuang H-C, Lay J-D, Hsieh W-C, Su I-J (2007) Pathogenesis and mechanism of disease progression from hemophagocytic lymphohistiocytosis to Epstein–Barr virus-associated T-cell lymphoma: nuclear factor-κB pathway as a potential therapeutic target. Cancer Sci 98:1281–1287
Jordan MB, Allen CE, Weitzman S et al (2011) How I treat hemophagocytic lymphohistiocytosis. Blood 118:4041–4052
Acknowledgments
This work was supported by the National Natural Science Fund (81270653), Beijing Natural Science Fund (7132087), Public Health Project of Science and Technology Committee of the Beijing Municipal Development projects (Z131100006813041), and Friendship Hospital research fund (yyqdkt2013-10).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jing-Shi, W., Yi-Ni, W., Lin, W. et al. Splenectomy as a treatment for adults with relapsed hemophagocytic lymphohistiocytosis of unknown cause. Ann Hematol 94, 753–760 (2015). https://doi.org/10.1007/s00277-014-2276-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-014-2276-9