
Abstract
We present an unusual case of a patient with extreme lymphoplasmacytosis and hepatic failure in association with a reaction to sulfasalazine and a concurrent Epstein-Barr virus (EBV) infection. Sulfa drugs can cause a wide range of allergic and hypersensitivity reactions and occasionally can lead to a fulminant illness. In the case under discussion the patient had hepatotoxicity, skin rash, fever, and peripheral blood atypical lymphocytosis. Initial impressions suggested the possibility of a malignant lymphoproliferative disorder. Flow cytometry of peripheral blood and a bone marrow biopsy provided clear evidence for a reactive, polyclonal process as opposed to a malignant disorder. Cessation of the offending drug and administration of steroids led to dramatic improvement. This case illustrates that drug hypersensitivity reactions can be manifested by an extreme lymphocytoid leukemoid reaction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reactions to sulfonamides are common and range from mild to fulminant systemic hypersensitivity reactions. These may include hepatotoxicity, skin rash, and fever. Occasionally, these reactions are also accompanied by a variable degree of lymphocytosis.
We report an unusual case of a patient with marked peripheral blood lymphoplasmacytosis associated with a reaction to sulfasalazine and a concurrent Epstein-Barr virus (EBV) infection. As this case and a review of the literature suggest, drug-induced hypersensitivity reactions with or without an associated viral infection should be included in the differential diagnosis of patients presenting with an extreme lymphocytosis. We are unaware of any published case of a drug reaction to sulfonamide associated with a white blood count >100,000/μl. This case illustrates the importance of recognizing that even an extreme lymphoplasmacytosis can occur as part of a hypersensitivity reaction emphasizing the importance of discriminating this from leukemia.
Case summary
The patient, a 34-year-old male from Vermont, was in good general health except for occasional mild episodes of presumed Crohn's disease diagnosed over 20 years ago. He developed a perirectal abscess in October 1998 and at that time was placed on sulfasalazine 1 g four times a day which was the only medication he was taking. He ran out of his sulfasalazine in November 1998 and resumed it on 11 December 1998. On 12 December 1998 he noted the development of a macular rash over his arms, later spreading to his trunk and lower extremities. He subsequently developed recurrent fevers to 103–104°F, moderate diarrhea, and diffuse abdominal pain. On 14 December 1998 he was admitted to a local hospital where he was found to have elevated liver function tests, hyperbilirubinemia, and an elevated white cell count of 15,400/μl (laboratory results shown in Table 1 and Table 2). At that time his sulfasalazine was stopped and broad-spectrum antibiotics were started. Work-up, including hepatitis serologies, Monospot test, and blood cultures were all negative. An abdominal computed tomography (CT) scan showed enlarged mesenteric and inguinal lymph nodes and a slightly enlarged spleen. Over the next 4 days his overall condition progressively worsened with rapidly rising bilirubin, abnormal liver function tests, leukocytosis, and a prolonged prothrombin time. He developed bloody diarrhea and a fall in his hematocrit to 21%, which necessitated blood and fresh frozen plasma transfusions. The patient became confused and developed episodes of hallucinations for which he was started on lactulose. On 19 December 1998 he was transferred to the Gastroenterology Service at our institution for further management of acute liver failure and for evaluation for a liver transplant.
On arrival at our institution he was found to be confused and lethargic. He had a fever of 101°F, a diffuse erythematous macular rash most pronounced over his abdomen and legs, diffuse shotty lymphadenopathy, and slight hepatosplenomegaly. The laboratory results were significant for a white cell count of 102,300/μl, with the following differential: 28% neutrophils, 51% lymphoid cells, 2% monocytes, 10% eosinophils; hematocrit (Hct) 21%; total bilirubin of 10.4 mg/dl; aspartate aminotransferase (AST) 573 IU/l; alanine aminotransferase (ALT) 1093 IU/l; NH3 138 μmol/l; and lactic dehydrogenase (LDH) 2010 IU/l. He was admitted to the Medical Intensive Care Unit and lactulose was continued. Cross-matching for blood transfusion was complicated by the presence of a cold autoantibody. A hematology consult was requested to distinguish between leukemoid reaction versus acute leukemia.
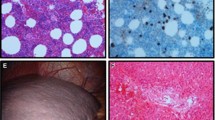
Review of his peripheral smear (Fig. 1) showed a marked increase in mononuclear cells of varying size and shape. Large cells with high nuclear/cytoplasmic ratio, plasmacytoid lymphocytes, and plasma cells, some binucleated, were noted. There were 10% eosinophils. Examination of the red blood cells showed prominent red cell agglutination, occasional nucleated red blood cells, and the platelet count was mildly decreased.

Peripheral blood smear (×400) demonstrates the presence of numerous circulating atypical lymphocytes, plasmacytoid cells and plasma cells (solid arrows) as well as eosinophils (b, dashed arrow). c Note the presence of prominent red cell agglutination (solid arrow) as well as rouleaux formation (dashed arrow)
A bone marrow biopsy was performed and demonstrated a normocellular marrow for age with an interstitial mononuclear cell infiltrate comprised of variable numbers of lymphocytes, plasmacytoid lymphocytes, and plasma cells, representing about 40–50% of the total nucleated cells (Fig. 2). An increased number of eosinophils was also seen. Otherwise erythroid, myeloid, and megakaryocytic hematopoiesis was preserved.

Bone marrow specimen. Bone marrow aspirate (×400) demonstrates a large number of plasma cells (a, solid arrows) as well as eosinophils (b, dashed arrows). c Bone marrow biopsy specimen (×400) shows the presence of a mononuclear infiltrate as well as a high number of eosinophils. Cellularity as well as erythroid, myeloid, and megakaryocytic maturation is preserved
The differential diagnosis at that time included drug reaction, mononucleosis-like viral syndrome, or a lymphoid malignancy. Interventions were withheld and further studies were requested. Flow cytometry of the peripheral blood showed that the majority of the circulating cells were polyclonal CD19-positive B cells based on surface light chain staining. A major subpopulation of the B cells lacked the B cell marker CD20 while coexpressing CD138, indicating plasma cell differentiation. The residual T cells were mainly activated cytotoxic CD8, HLA-DR-positive cells. A skin biopsy was also performed and showed papillary dermal edema and a superficial lymphohistiocytic and eosinophilic infiltrate suggestive of a hypersensitivity reaction, consistent with early erythema multiforme. Further laboratory work-up showed hypocomplementemia and hypofibrinogenemia (Table 2). His serum viscosity was normal. His serologies [including human immunodeficiency virus (HIV)] were all negative except for a positive Monospot test as well as IgG and IgM antibodies directed against EBV viral capsid antigens. Serum protein electrophoresis showed diffuse hypergammaglobulinemia without any monoclonal spikes.
Methylprednisolone 40 mg twice a day was started on 20 December 1998 followed by rapid clinical improvement and normalization of his laboratory studies. By 22 December 1998 his white cell count decreased to 15,800/μl and his liver functions became normal. He was discharged in good overall condition on 26 December 1998 and was seen in follow-up in January 1999. Clinically he was fully recovered and all his laboratory tests were within normal limits. The EBV serologies showed a positive viral capsid IgG and negative IgM titers.
Discussion
In this report we present the case of a systemically ill patient with hepatic failure and a rapidly progressive, extreme lymphocytosis masquerading as an acute hematological malignancy. The temporal association, constellation of clinical and pathological findings as well as the rapid improvement after cessation of the suspected offending drug and initiation of steroid treatment suggested a drug reaction as the etiology of this patient's illness.
Sulfa drugs are frequently associated with allergic reactions and can cause hypersensitivity reactions ranging from mild symptoms to serious side effects such as Stevens-Johnson syndrome and agranulocytosis. There have been several reports of a unique mononucleosis-like hypersensitivity syndrome due to sulfa drugs consisting of hepatitis, fever, rash, and atypical lymphocytosis similar to our patient's case.
Sulfa drugs have been associated with lymphocytosis or lymphoplasmacytosis with varying numbers of atypical lymphocytes, in peripheral blood and/or bone marrow. As early as the 1950s there were reports describing bone marrow plasmacytosis along with fever, skin rash, and hepatitis associated with the use of sulfonamides [1, 2]. Similar hypersensitivity reactions have been described in association with other drugs, most frequently antiepileptic agents such as phenytoin, phenobarbital, and carbamazepine [3]. Bocquet et al. [4] suggested the acronym DRESS syndrome (drug rash with eosinophilia and systemic symptoms) in order to distinguish this particular syndrome from other hypersensitivity reactions.
A review of the literature describes several reports of patients taking sulfa drugs with a mononucleosis-like picture and lymphoplasmacytosis (Table 3) [5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18]. The clinical diagnosis in these patients include inflammatory bowel disease, both Crohn's disease, as in our patient, and ulcerative colitis, psoriasis, dermatitis herpetiformis, and arthritis—not surprisingly diseases frequently treated with long-term administration of sulfa drugs. Interestingly, the majority of the patients were young with a range of age from 19–45 and a mean age of 30 years. Most cases occurred within 2–5 weeks after the start of sulfa treatment. Most patients improved with withdrawal of the sulfa drug and/or use of steroids. One patient died from fulminant hepatitis and pancreatitis felt to be related to his sulfasalazine treatment despite steroid treatment [5]. Interestingly, two patients had recurrences [6, 7]; in one patient symptoms recurred after she unintentionally received cotrimoxazole (trimethoprim-sulfadiazine) suggesting that the syndrome was due to the sulfa component of sulfasalazine. Our patient who also fits the age category of previously described cases took sulfasalazine for a few weeks then discontinued it and his symptoms appeared only a few days after resuming sulfasalazine, similarly suggesting prior sensitization to the drug as one of the contributing factors.
The majority of patients had white blood counts below 50,000/μl, with only two patients having a true lymphocytic leukemoid reaction (70 and 90,000/μl). None of the patients had lymphocytosis to the degree observed in the case of our patient. The degree of lymphocytosis as well as the percentage of lymphocytes, atypical lymphocytes, plasmacytoid lymphocytes, and plasma cells was variable; notably, two cases had increased circulating plasma cells (9 and 35%). In addition to the lymphoplasmacytosis, and as might be expected in a drug reaction, eosinophilia was seen in four patients. Our patient had a lymphoplasmacytosis with 28% atypical lymphocytes and 16% plasma cells. Eosinophilia was seen in the peripheral blood as well as in the bone marrow. Four patients had bone marrow biopsies performed that showed variable degrees of lymphoplasmacytosis and eosinophilia.
Flow cytometry immunophenotypic data were available in two cases and showed that the cells were predominantly activated T cells [7, 17]. In contrast, in our case flow immunophenotyping demonstrated that the majority of cells in the peripheral blood were polyclonal B cells and plasma cells, with a minor population of activated T cells. Within the T cell population the CD4/8 ratio was 0.56. While flow cytometry was not performed in other cases of sulfasalazine-induced hypersensitivity reactions, plasmacytosis and the presence of lymphoplasmacytoid cells have been described in several cases suggesting that the flow cytometry findings in our case are not unusual.
Our patient also had evidence of a concurrent acute EBV infection corroborated by a positive IgM titer on presentation that became negative on follow-up. Acute infectious mononucleosis in itself can lead to an acute illness similar to our patient's case—fever, atypical lymphocytosis, and hepatitis. The extreme degree of lymphocytosis not previously described with EBV infection, peripheral blood and bone marrow eosinophilia as well as the skin biopsy being consistent with a drug reaction suggest that EBV infection could not be the sole explanation for our patient's illness. Also, in acute EBV infection the atypical lymphocytes are predominantly activated cytotoxic T cells as opposed to B cells. There was no evidence of EBV infection in the bone marrow specimen by immunohistochemistry for latent membrane protein.
Our case suggests that the marked lymphoplasmacytosis was due to both a reaction to a sulfa drug and a concurrent viral infection (infectious mononucleosis). Both conditions have an underlying immunologic mechanism and therefore one may speculate that the marked lymphoplasmacytosis may represent a "cumulative effect" from both conditions. This case also shows the importance of considering a reactive condition in the differential diagnosis of a patient presenting with an extreme white blood count in order to avoid unnecessary and potentially toxic treatments. Both bone marrow biopsy and flow cytometry immunophenotyping may be helpful in confirming the nature of the elevated count. Prompt cessation of the offending drug is critical to avoid irreversible organ damage. Anecdotal evidence in this and other patients' cases suggests that administration of steroids may be beneficial and can lead to rapid clinical improvement.
References
Wolf J, Worken B (1954) Atypical amyloidosis and bone marrow plasmacytosis in a case of hypersensitivity to sulfonamides. Am J Med 16:746–755
Tisdale WA (1958) Focal hepatitis, fever and skin rash following therapy with sulfamethoxypyridazine, a long-acting sulfonamide. N Engl J Med 258:687–690
Callot V, Roujeau J-C, Bagot M, Wechsler J, Chosidow O, Souteyrand P, Morel P, Dubertret L, Avril M-F, Revuz J (1996) Drug-induced pseudolymphoma and hypersensitivity syndrome. Arch Dermatol 132:1315–1321
Bocquet H, Bagot M, Roujeau J-C (1996) Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg 15:250–257
Rubin R (1994) Sulfasalazine-induced fulminant hepatic failure and necrotizing pancreatitis. Am J Gastroenterol 89:789–791
Han T, Chawla PL, Sokal JE (1969) Sulfapyridine-induced serum-sickness-like syndrome associated with plasmacytosis, lymphocytosis and multiclonal gamma-globulinopathy. N Engl J Med 280:547–548
Gyssens IC, De Bock RF, Peetermans ME (1989) Sulfasalazine allergy: fever, skin rash, hepatitis and T-lymphocytosis. Ned Tijdschr Geneeskd 133:1608–1610
Sotolongo RP, Neefe LI, Rudzki C, Ishak KG (1978) Hypersensitivity reaction to sulfasalazine with severe hepatotoxicity. Gastroenterology 75:95–99
Kromann NP, Vilhelmsen R, Stahl D (1982) The dapsone syndrome. Arch Dermatol 118:531–532
Poland GA, Love KR (1986) Marked atypical lymphocytosis, hepatitis and skin rash in sulfasalazine drug allergy. Am J Med 81:707–708
Pettersson T, Gripenberg M, Molander G, Friman C (1990) Severe immunological reaction induced by sulphasalazine. Br J Rheumatol 29:239–240
Leroux JL, Chertok P, Ghezail M, Blotman F (1992) Hypersensitivity reactions to sulfasalazine: skin rash, fever, hepatitis and activated lymphocytes. Clin Exp Rheumatol 10:427
Mihas AA, Goldenberg DJ, Slaughter RL (1978) Sulfasalazine toxic reactions. Hepatitis, fever and skin rash with hypocomplementemia and immune complexes. JAMA 239:2590–2591
Tohyama M, Yahata Y, Yasukawa M, Inagi R, Urano Y, Yamanishi K, Hashimoto K (1998) Severe hypersensitivity syndrome due to sulfasalazine associated with reactivation of human herpesvirus 6. Arch Dermatol 134:1113–1117
Hautekeete ML, Bourgeois N, Potvin P, Duville L, Reynaert H, Devis G, Adler M, Kloppel G (1992) Hypersensitivity with hepatotoxicity to mesalazine after hypersensitivity to sulfasalazine. Gastroenterology 103:1925–1927
Labadie H, Beaugrand M, Ferrier JP (1986) Hepatitis and mononucleosis-like syndrome associated with salazosulfapyridine. Rev Med Interne 7:35–40
Iwatsuki K, Tsugiki M, Tagami H, Yamada M (1984) Infectious mononucleosis-like manifestations. An adverse reaction to sulfasalazine. Arch Dermatol 120:964–965
Foresti V, Tronci M, Ungaro A, Confalonieri F (1988) Severe sulfasalazine-induced hypersensitivity reaction simulating mononucleosis. Recenti Prog Med 79:382–383
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Halmos, B., Anastopoulos, H.T., Schnipper, L.E. et al. Extreme lymphoplasmacytosis and hepatic failure associated with sulfasalazine hypersensitivity reaction and a concurrent EBV infection—case report and review of the literature. Ann Hematol 83, 242–246 (2004). https://doi.org/10.1007/s00277-003-0746-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-003-0746-6