
Abstract
The Raman and Cr3+ and V2+ luminescence spectra of beryl and emerald have been characterized up to 15.0 and 16.4 GPa, respectively. The Raman spectra show that an E 1g symmetry mode at 138 cm−1 shifts negatively by −4.57 (±0.55) cm−1/GPa, and an extrapolation of the pressure dependence of this mode indicates that a soft-mode transition should occur near 12 GPa. Such a transition is in accord with prior theoretical results. Dramatic changes in Raman mode intensities and positions occur between 11.2 and 15.0 GPa. These changes are indicative of a phase transition that primarily involves tilting and mild distortion of the Si6O18 rings. New Raman modes are not observed in the high-pressure phase, which indicates that the local bonding environment is not altered dramatically across the transition (e.g., changes in coordination do not occur). Both sharp line and broadband luminescence are observed for both Cr3+ and V2+ in emerald under compression to 16.4 GPa. The R-lines of both Cr3+ and V2+ shift to lower energy (longer wavelength) under compression. Both R-lines of Cr3+ split at ~13.7 GPa, and the V2+ R1 slope changes at this pressure and shifts more rapidly up to ~16.4 GPa. The Cr3+ R-line splitting and FWHM show more complex behavior, but also shift in behavior at ~13.7 GPa. These changes in the pressure dependency of the Cr3+ and V2+ R-lines and the changes in R-line splitting and FWHM at ~13.7 GPa further demonstrate that a phase transition occurs at this pressure, in good agreement with our Raman results. The high-pressure phase of beryl appears to have two Al sites that become more regular under compression. Hysteresis is not observed in our Raman or luminescence spectra on decompression, suggesting that this transition is second order in nature: The occurrence of a second-order transition near this pressure is also in accord with prior theoretical results. We speculate that the high-pressure phase (beryl-II) might be a mildly modulated structure, and/or that extensive twinning occurs across this transition.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
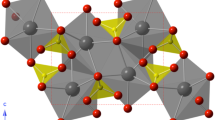
Beryl is a beryllium ring silicate that crystallizes in the hexagonal crystal system with P6/mcc symmetry (Fig. 1). Emerald (Be3Al2Si6O18:Cr) is a chromium-bearing variety of beryl, and most emerald samples also contain hundreds to thousands of ppm of vanadium (Ollier et al. 2015). Ring silicates have attracted considerable interest due to their unusual thermal expansion behavior, as well as the ability for their zeolite-like channels to contain a wide range of volatiles (e.g., H2O and CO2). For example, the structurally related ring silicate cordierite ((Mg,Fe)2Al4Si5O18) on being heated to ~800 °C expands along the a-axis, but contracts along the c-axis (Milberg and Blair 1977; Hochella and Brown 1986). The high-temperature behavior of beryl is comparable: When heated to ~800 °C, it behaves much like cordierite. Below ~300 °C, the c-axis contracts with increasing temperature, but expands at higher temperatures (Morosin 1972; Fan et al. 2015). Interestingly, the high-temperature behavior of emerald shows a shallower minimum in the c-axis contraction with temperature, and the minimum distance is shifted to temperatures of ~100 °C by only 0.5 wt% Cr substitution into the Al3+ sites (Morosin 1972). This has been attributed to expansion of the unit cell of emerald relative to end-member beryl, since Cr3+ has a larger ionic radius than Al3+. Ring silicates such as cordierite have widespread industrial uses as ceramic materials, but the industrial applications of beryl have been more limited, and are usually highly specialized due to the toxicity associated with beryllium (Hochella and Brown 1986). Emerald has attracted more interest as a tunable laser material (Buchert et al. 1983; Lai 1987) and has also been mined as a gem since before the start of the Egyptian Ptolemaic Kingdom.

Crystal structure of beryl looking down the c-axis. Structure after Gibbs et al. (1968). Dashed line indicates the unit cell. Crystal structure diagram generated using v8.7.6 CrystalMaker®
The high-pressure behavior of ring silicates such as cordierite and tourmaline has also attracted interest due to their occurrence in subduction zone environments and their widespread industrial uses (Likhacheva et al. 2012; Miletich et al. 2014a, b; Finkelstein et al. 2015; Xu et al. 2016). Experimental high-pressure investigations of beryl are limited to X-ray diffraction studies and two luminescence studies. Early high-pressure single-crystal work on a synthetic chromium-bearing beryl sample by Hazen et al. (1986) extended to ~5.7 GPa. Hazen et al. (1986) reported high-pressure structures up to a maximum pressure of 5.7 GPa and did not observe any phase transitions. Qin et al. (2008) conducted high-pressure powder diffraction on natural beryl up to ~19.2 GPa and did not observe a phase transition, but the composition of their sample is unknown. More recently, Fan et al. (2015) conducted both powder diffraction to 16.7 GPa and single-crystal diffraction to 9.5 GPa on a natural colorless gem-quality beryl sample of unknown composition. They did not report high-pressure crystal structures and also did not observe a phase transition. Prencipe and Nestola (2005) conducted single-crystal high-pressure X-ray diffraction on natural colorless beryl to ~6.3 GPa in order to obtain accurate unit cell parameters as a check on their theoretical calculations. Two previous high-pressure studies of Cr3+ luminescence spectra of emerald up to 8.0 and 5.0 GPa report pressure-induced red shifts of the main R-line (Canny et al. 1987; Sánchez-Alejo et al. 2011). Sánchez-Alejo et al. (2011) report a difference in the R-line shift of ~20 % between natural and synthetic samples of emerald. To our knowledge, high-pressure vibrational spectroscopic studies of beryl have not been reported. On the theoretical side, ab initio calculations on high-pressure, high-temperature behavior and the mechanics of the compression of beryl have been conducted (Prencipe and Nestola 2005, 2007; Prencipe et al. 2011). Prencipe et al. (2011) predict that a second-order, soft-mode-induced phase transition occurs at ~14 GPa, with a lowering of symmetry from hexagonal P6/mcc to triclinic P \(\bar{1}\). High-pressure single-crystal measurements indicate that the symmetry lowers to triclinic at pressures near 14 GPa and twinning occurs (M. Merlini, personal communication).
We utilize the fluorescence of chromium and vanadium to probe the high-pressure behavior of natural emerald. Investigations of the high-pressure behavior of Cr3+ or V2+ luminescence have largely been restricted to relatively simple oxides and rare earth-bearing garnets (Chopelas and Nicol 1982; Kottke and Williams 1983; Wamsley and Bray 1994; Jovanić 1997, 2000; Jovanić et al. 2010). Of these, the most well-characterized material is ruby (Al2O3:Cr), whose role as a pressure calibrant is well known (Piermarini et al. 1975; Mao et al. 1986). Ruby is also the model system through which much of crystal field theory was developed (Sugano and Tanabe 1958; Burns 1993). The materials that have had their luminescent behavior probed under pressure share a few general properties: They are simple oxide systems, they are not hydrated, and they all have Al sites that are close to ideal octahedra. High-pressure investigations of Cr3+ luminescence from more distorted Al sites in silicates and oxides are limited (e.g., O’Bannon and Williams 2016). And, high-pressure studies of the simultaneous emission of two or more luminescent centers in oxides have been largely confined to studies on charge transfer between transition metal elements and rare earth elements in garnets (Shen et al. 2000).
Since both Cr3+ and V2+ have a d 3 configuration, their luminescence spectra are quite similar. The main differences lie in the intensity, wavelength, and lifetime of emission associated with these transition metals. The emission of Cr3+ and V2+ is typically characterized by relatively narrow intense R-lines associated with the spin-forbidden 2E–4A2 transition, with the 4A2 state being the ground state and the 2E state being split in non-cubic environments (Syassen 2008). At high field strengths, the narrowband 2E−4A2 emission typically dominates the fluorescence spectrum, while at low crystal field strengths the generally broader (and spin-allowed) 4T2–4A2 emission dominates the emission spectrum, and at intermediate field strengths, both can occur together (Sugano and Tanabe 1958; Burns 1993; Gaft et al. 2005). In samples with significant Cr3+ or V2+ substitution, sharp neighbor lines (N-lines) generated by shared excitations between neighboring Cr3+ or V2+ centers can also be observed. These bands tend to be complex, since a wide variety of potential neighbor interactions exist (especially in complex crystal structures). If their assignments are known, their pressure dependence can be used to estimate changes in magnetic interactions between chromium centers (Williams and Jeanloz 1985). In addition to these electronic transitions, vibronic bands are also observed. These phonon-associated sidebands are offset from the R-lines by the frequency of a lattice phonon or a local vibrational mode.
Here, we use Raman spectroscopy to probe the bonding environment of beryl up to ~15 GPa, and Cr3+ and V2+ luminescence to probe the Al site in emerald (Be3Al2Si6O18:Cr,V) under compression up to 16.4 GPa. The Cr3+ luminescence spectrum of emerald under compression has been previously reported but was characterized only to ~5.0 GPa (Sánchez-Alejo et al. 2011). They reported solely the pressure dependence of the R1 line, although the emission spectrum also contains the Cr3+ 4T2 emission, the R2 band, linewidths, and, for some compositions, V2+ emission. With respect to the vibrational spectra, high-pressure Raman spectroscopic studies on beryl appear not to have been previously conducted. Thus, our experiments are oriented toward: (1) probing the local bonding environment and of the Si6O18 rings and Al/Be sites to constrain their high-pressure behavior, (2) resolving the 2E and 4T2 emission bands from both Cr3+ and V2+ in the same crystal structure under compression, (3) determining whether any phase transition occurs, such as the soft-mode transition that has been proposed to be present near ~14 GPa as predicted by theory (Prencipe et al. 2011), (4) determining how the crystal field strength in emerald changes under compression, and (5) examining how the symmetry of the Cr-substituted Al sites might change under compression by examining shifts in splitting and linewidths of luminescence peaks.
Experimental methods
The beryl samples are from Rabenstein, Zwiesel, Lower Bavaria, Germany (UCSC sample #6533), and the emerald samples are from the Boyacá Department, Columbia. Purity of both beryl and emerald samples was confirmed by single-crystal X-ray diffraction, Raman, infrared, and luminescence spectroscopy. Spectroscopic measurements were in good agreement with previous studies (Wood and Nassau 1967; Hofmeister et al. 1987; Hagemann et al. 1990; Sánchez-Alejo et al. 2011). Moreover, both samples consisted of gem-quality euhedral single crystals with typical beryl crystal habit. The beryl sample is light green in color, while the emerald sample is a characteristic emerald/deep green color. The beryl sample was selected for the Raman measurements due to its low luminescence background, and the emerald sample was selected for the relatively high amplitude of its narrowband (R-line) emission relative to broadband emission.
Blocks of the beryl and emerald with dimensions of ~0.05 × 0.05 × 0.02 mm3 were mounted on the goniometer head of a Stoe Stadi-Vari diffractometer equipped with an RDI CMOS1 M detector on beamline 12.2.2 at the Advanced Light Source in Berkeley, CA. Diffraction data were collected using synchrotron radiation monochromated with silicon(111) to a wavelength of 0.4959(1) Å (25 keV). A hemisphere of data was collected using 0.25° Ω scans at multiple χ positions with 0.5-s exposure times. Data were indexed and reduced using the X-Area program. Unit cell parameters and volumes obtained are listed in Table 1. Hence, based on the systematics of the variations of the a-axis dimension with c/a ratio of Aurisicchio et al. (1988), each of these beryls are considered “normal” beryls with limited substitution of divalent cations for aluminum, and limited lithium substitution.
Trace element concentrations of the beryl and emerald were measured with a PhotonMachines Analyte 193H, which is a 193-nm ArF excimer laser system coupled with a ThermoScientific ElementXR single-collect or magnetic sector ICP-MS. The instrument was calibrated with a SRM 610 trace element glass from NIST, and Al was used as the internal standard. The results are given in Table 2. The concentrations of Na, K, Mg, Ca, and Li are similar in the two beryl crystals that we study, and their lattice parameters are also similar (indeed, the c/a ratios are identical, see Table 1). Thus, we do not anticipate that the high-pressure behavior of these two samples will be notably different from one another. In passing, we note that the high-pressure behavior of alkali-rich beryls is likely different from that of alkali-poor beryls. Alkali-rich beryls show a lengthening of the lattice parameters and larger unit cell volumes (due to alkali substitution) relative to alkali-poor beryls (Sherriff et al. 1991), and the role of alkali occupancy of channels within beryls or lithium substitution generating beryllium vacancies (Aurisicchio et al. 1988) on high-pressure transitions is beyond the scope of this study.
High static pressures were generated using a Merrill–Bassett-type diamond anvil cell (DAC) equipped with 16-sided type 1a 500-µm culet diamond anvils. A spring steel gasket with a 200-µm hole was used as the sample compartment. Experiments were carried out with a methanol/ethanol 4:1 mixture as the pressure media, which yields hydrostatic conditions up to ~10.5 GPa (Piermarini et al. 1973; Fujishiro et al. 1982; Klotz et al. 2009). There is a pressure gradient in our sample compartment above ~10.5 GPa. However, since our spectra were collected from a spot size of ~2 μm in diameter, we do not anticipate that pressure variations will adversely affect our spectra; moreover, at our maximum pressure of 16.4 GPa, deviations of pressure in methanol/ethanol are anticipated to be of order 1 GPa across 150- to 200-µm sample compartments (Klotz et al. 2009). Single crystals of both the sample and ruby were loaded into the sample compartment, and to prevent contamination of the sample luminescence spectra by the intense ruby luminescence, the small ruby crystals (ca. 2 μm diameter) and the emerald were spatially separated within the sample. The standard ruby fluorescence method was used to determine the pressure (Mao et al. 1986). For the low-temperature, ambient-pressure measurements, the sample was immersed in liquid nitrogen.
High-pressure Raman spectra were collected using a 600 lines/mm grating and 633-nm excitation between 75 and 1200 cm−1, while high-pressure luminescence spectra used a 1800 lines/mm grating with 532-nm excitation from 650 to 800 nm (15,380–12,500 cm−1). Both Raman and luminescence spectra were measured using a Horiba LabRAM HR Evolution Raman spectrometer with a spectrometer focal length of 800 mm. An Olympus BXFM-ILHS microscope with a 50× long working distance objective was used to focus the laser beam onto the sample. Raman spectra were collected to a pressure of ~15 GPa, and luminescence spectra were collected to a pressure of ~16.4 GPa and on decompression to room pressure at 298 K. Infrared spectra were collected from 400 to 8500 cm−1 using a Bruker Vertex 70v evacuated Fourier transform infrared spectrometer (FTIR) equipped with a globar source, KBr beam splitter, and a liquid-N2-cooled mercury–cadmium–telluride (MCT) detector. Infrared spectra have a resolution of 4 cm−1. Vibrational and luminescence spectra were fit using a combination of Gaussian and Lorentzian functions with either Horiba Labspec6 software or Bruker OPUS6.5 software.
Results and discussion
Room-pressure luminescence and vibrational spectra
Raman spectra of beryl and emerald under 532-nm excitation are shown in Fig. 2a and are in excellent agreement with previous studies (Wood and Nassau 1967; Hofmeister et al. 1987; Hagemann et al. 1990). Due to the broad luminescence background of Cr3+ and V2+ emission, the Raman active H2O modes in the ~3600 cm−1 range of emerald could not be resolved. Mid-infrared absorption spectra of the emerald show that both type I water and II water are present within the channels (e.g., Wood and Nassau 1967). H2O combination bands and the first overtone of H2O are also present in the spectra (Fig. 2b). The Raman spectra of the OH region in beryl did not have interference from luminescence, and type I and II water is observed in the channels (Fig. 2b inset). Very weak Raman modes are observed at ~1385 cm−1 in both beryl and emerald and are due to CO2 contained in the channels. Assignments and peak positions of observed Raman modes are given in Table 3.

Room-pressure and room-temperature a Raman spectra of beryl and emerald in the lattice- and ring-vibration region and b infrared spectrum of emerald in the OH stretch region. Inset shows Raman spectrum of beryl in the OH stretch region
The assignments and deconvolution of the emission bands in emerald at room pressure and temperature under 532-nm excitation are shown in Fig. 3 and listed in Table 3. In accord with prior work (e.g., Kisliuk and Moore 1967; Schmetzer and Eysel 1974; Ollier et al. 2015), we assign the sharp narrow emission bands to the transition from the 2E states to the ground states of Cr3+ and V2+: These are typically referred to as the R-lines. The splitting of the 2E states into two components (R1 and R2) is a consequence of the site symmetry of Al in the beryl lattice being slightly distorted from octahedral: Its site symmetry is D 3 (e.g., Gibbs et al. 1968). The broad emission bands are assigned to the transition from the 4T2 states to the ground states of Cr3+ and V2+. The Cr3+ 4T2 emission band’s location at 702.3 nm (14,239 cm−1) at room pressure and temperature is at substantially longer wavelength than the corresponding absorption of this transition near 615 nm (Kisliuk and Moore 1967). Thus, the excited state of this transition is Stokes shifted to the long wavelength side of the R-lines, a phenomenon that has been well documented in emerald (Kisliuk and Moore 1967). The 4T2 emission band of V2+, located at 731.55 nm (13,669.6 cm−1), is also Stokes shifted to longer wavelengths relative to its absorption band at ~667 nm (~15,000 cm−1) (Schmetzer and Eysel 1974). Our emerald sample is grossly similar in Cr3+ and V2+ concentration to the Capoeirana, Brazil emerald sample from Ollier et al. (2015), and we fit and assign our spectra similarly to their ambient-pressure study. The previous assignments of the V2+ 2E and 4T2 emission lines are based on lifetime measurements (Schmetzer and Eysel 1974; Ollier et al. 2015).

Typical deconvolution of emerald emission spectrum using 532-nm excitation. Assignment of emission bands based on Schmetzer and Eysel (1974) and Ollier et al. (2015). Green colored peaks are Cr3+ emission and wine colored peaks are V2+ emission. Inset shows detailed 2E fit with primary (p) and secondary (s) R1 and R2 lines. The modulation/fringing on the broad 4T2 emission is an artifact
Unlike Ollier et al. (2015), we observe both the R1 and R2 lines from Cr3+ under steady-state excitation (they observe the R2 band only in time-delayed spectra), and we fit two components to each Cr3+ R-line (Fig. 3 inset). The lower-intensity component/shoulder of each R-line is located to the long wavelength side of the more intense component of the R-lines, and the presence of these additional components suggests that there are likely two modestly different Cr3+ sites, with most of the Cr3+ being present in the site represented by the more intense bands. We refer to the most intense component of the Cr3+ R-lines as the primary R-lines and the lower-intensity components as the secondary R-lines. The origin of the secondary Cr3+ R-lines might be associated with local shifts in bonding associated with channel constituents. Certainly, the shift in energy of the secondary lines from the primary R-lines is slight, and water as a channel constituent affects the bonding of the adjoining silica tetrahedra (e.g., Fridrichová et al. 2016), and hence proximity to a water molecule in a channel may modestly alter the charge density of the Al/Cr site and give rise to the secondary bands. Peak asymmetries, and the necessity of fitting absorption bands with two peaks of markedly different intensities, have also been noted in the absorption spectrum of emerald (Parkin and Burns 1980; Taran et al. 1994), and additional R-lines from separate Cr sites (albeit on the short-wavelength side of the primary R-lines) have been observed in very Cr-rich emerald (3 at.%, Quarles et al. 1988).
The tandem of Cr3+ and V2+ can account for all of the emission bands observed, except for the broad low-intensity band located at ~785 nm. Ollier et al. (2015) discuss the possibility that this broad low-intensity band might be due to the 2E to 2T2 transition of Ti3+, but note that this valence state is unlikely in geologic materials. They suggest that emission from ferric iron is the probable origin, due to the large full width at half maximum (FWHM) of the ~785-nm emission band. Moreover, a broad Fe3+ emission band near 770 nm has been reported in ruby doped with both Cr3+ and Fe3+ (Snytnikov et al. 2008). Hence, we tentatively assign the emission band at ~785 nm observed in our room-pressure and room-temperature measurements to Fe3+, since the Ti concentration in our sample was below detection limits. Under compression, the intensity of this band decreases dramatically and becomes unresolvable, so the deconvolutions of our high-pressure spectra do not include this emission band.
The 77 K luminescence spectrum of emerald is shown in Fig. 4. At 77 K and room pressure, the 2E transition of Cr3+ and V2+ and pair emission lines of Cr3+ and V2+ are the primary features in the spectrum. At 77 K, the Cr3+ R-lines shift to lower wavelength by ~1 nm and the splitting between the primary R1 and R2 is lowered to 62.9 cm−1 (from 65.8 cm−1 at room temperature), which is in accord with previous studies (Ohkura et al. 1987; Skvortsova et al. 2015). At low temperature (77 K), the broad thermally populated 4T2 emissions of Cr3+ and V2+ are quenched. The secondary R-lines are still observed at 77 K (inset in Fig. 4), indicating that they are not populated by thermal effects and supporting their origin as being derived from a separate Cr3+ site. Peak positions of Cr3+ neighbor lines are assigned after Ohkura et al. (1987) and shown in Fig. 4 (dashed vertical lines). Neighbor lines (N-lines) are emission lines that are generally located to the longer wavelength side of the R-lines (lower energy) and are attributed to paired ion emissions. Interestingly, the previously assigned neighbor lines account for less than half of the observed sharp emission lines in the sideband region (indeed, many bands were not assigned by Ohkura et al., 1987). This demonstrates that this region consists of complex pair emission lines due to neighbor interactions from Cr3+ and probably V2+-associated pairs.

Representative room-pressure 77 K luminescence spectra of emerald using 532-nm excitation. The sharp emission lines in the ×50 spectrum at low temperature are assigned to paired emissions of Cr and/or V (N-lines). Inset shows detailed 2E fit with primary (p) and secondary (s) R1 and R2 lines. Vertical red dashed lines are the subset of Cr N-lines that were assigned by Ohkura et al. (1987)
High-pressure Raman spectra
Representative high-pressure Raman spectra are shown on compression and decompression in Fig. 5, and the pressure shifts of the Raman modes are shown in Fig. 6. The assignments, pressure shifts, and mode Grüneisen parameters (γ i ) of beryl are given in Table 3. The assignments of the Raman modes of beryl have attracted interest (Hofmeister et al. 1987; Hagemann et al. 1990; Prencipe et al. 2006): For reference, there are 36 Raman active modes of beryl predicted by factor group analysis (e.g., Prencipe et al. 2006). Most of the modes resolved in this study are assigned to either Si/Be–O stretching or bending vibrations, or ring breathing/distortions. Notably, the E 1g mode located at 138 cm−1 has a rapid negative shift under compression. However, we do not resolve this band above ~5 GPa due to our lower-frequency cutoff of ~100 cm−1. Nevertheless, its negative pressure shift appears to become larger with increasing pressure, and a polynomial extrapolation of the pressure shift of this mode (Fig. 6a) shows good agreement with the calculated mode shifts reported by Prencipe et al. (2011). This simple extrapolation of our Raman data to zero frequency thus suggests that the soft-mode transition would occur near 12 GPa. For comparison, the calculations of Prencipe et al. (2011) indicated that the decrease in frequency of this soft mode should induce a second-order transition near 14 GPa. However, the pressure shift of this E 1g mode likely becomes complicated as the transition is approached, with anharmonic effects becoming increasingly important, and the soft-mode frequency itself never reaching zero. Indeed, it likely approaches a minimum where the harmonic component of the effective potential is zero, and anharmonic effects predominate (Prencipe et al. 2011; Prencipe, personal communication). Hence, our polynomial extrapolation of the transition pressure may underestimate the actual pressure of the transition. Thus, given the pressure (and hence frequency) range over which we resolve this band, the agreement with theory (Prencipe et al. 2011) on both the shift and inferred pressure of a soft-mode transition in beryl/emerald is excellent (Fig. 6a). The other Raman modes shift monotonically and positively up to ~11.2 GPa, and at 15.0 GPa, all observed Raman modes undergo discontinuous shifts, dramatic decreases in intensity and broadening. On decompression, we do not observe hysteresis in the transition, although our bounds on the precise pressure of the transition from the Raman spectra are not tight.

High-pressure Raman spectra of beryl under 633-nm excitation on a compression and b decompression. Asterisks indicate the position of Cr3+ R-lines in the beryl sample

Raman mode shifts of a the low-frequency E 1g mode, including from the calculation of Prencipe et al. (2011) and b the higher-frequency lattice and silicate ring and Be modes. Closed symbols are compression, open symbols are decompression, and error bars are smaller than the symbols
We assign our modes in more detail using the calculated isotopic shifts of the Raman modes (Prencipe et al. 2006) as well as animations (animations are available on the CRYSTAL Web site)Footnote 1 of the Raman active modes (Prencipe et al. 2011). The Si and Be bending modes at 440, 520, and 678 cm−1 have variable pressure shifts; of these, the 678 cm−1 mode has the largest degree of Be motion, while the 520 cm−1 band predominantly involves Si and O motion, and the 440 cm−1 mode involves both Si and Al motion in addition to O, and its displacement appears to involve more of a bulk rotation of the silica tetrahedron (Prencipe and Nestola 2007, Prencipe et al. 2011). The smaller pressure shift of the 440 cm−1 band relative to the other two bands suggests that the internal tetrahedral deformational vibrations stiffen more rapidly under pressure than deformational vibrations associated with the rings: In this sense, the behavior of the 440 cm−1 band may more closely resemble that of the ring distortion and ring deformation vibrations at 318 and 391 cm−1. Interestingly, the pressure shifts of the Si–O2 stretching and O2–Al/Be–O2 deformational vibrations at 1005 and 1061 cm−1 are quite rapid, indicating that vibrations associated with the O2 atom are particularly responsive to pressure. The idea that the O2 atom may play a key role in the high-pressure deformational behavior of beryl is precedented: Prencipe and Nestola (2007) note that the mechanics of compression of beryl primarily involve movement of the O1 and O2 atoms.
While we could detect no evidence of Cr3+ luminescence from this beryl under 532-nm excitation, under 633-nm excitation low-intensity Cr3+ R-lines are observed (Fig. 5a, b). This indicates that the ability to excite the R-lines in Cr-poor beryl is minimal at 532 nm, while at 633 nm the strong 4A2g → 4T1g derived absorption band pumps the R-lines. Within Cr-rich emerald, the high-energy tail of this absorption band is sufficient to excite the R-line fluorescence. The emission lines in beryl are observed as contaminants in the Raman spectrum across the pressure range of this study and their pressure shifts, as described below, are essentially identical to the pressure shift observed in our emerald sample. The pressure shift of R1 in our beryl sample is −10.5 (±0.5) cm−1/GPa and for R2 is −10.1 (±0.3) cm−1/GPa. A more complete discussion of emerald Cr3+ R-line pressure shifts follows the next section.
Mode Grüneisen parameters
Using the pressure shift of a vibrational frequency ν i for a mode i, (dν i /dP), a mode Grüneisen parameter, or mode gamma (γ i ), can be calculated using γ i = K 0/ν 0i (dν i /dP). Here, K 0 is the bulk modulus of beryl, for which we used a value of 180 (±2) GPa (Fan et al. 2015). Two previous studies reported indistinguishable values of K 0 of 179 (±1) and 181 GPa (Yoon and Newnham 1973; Prencipe and Nestola 2005), while Hazen et al. (1986), in their single-crystal study to 5.7 GPa, reported a value of K 0 of 170 (±5). The ν 0i values were determined by Raman spectroscopy at room temperature and pressure (Fig. 2), and the calculated dν i /dP and mode gammas are listed in Table 1. Notably, the E 1g mode at 138 cm−1 has a large negative mode Grüneisen parameter, which is in good accord with the calculated Grüneisen parameters reported by Prencipe et al. (2011). For comparison, the thermodynamic value of the Grüneisen parameter can be determined using the thermal expansion reported by Morosin (1972), coupled with the bulk modulus of 180 GPa (Fan et al. 2015) and the heat capacity at 300 K (Hemingway et al. 1986) using γ i = α K 0/ρC p. The calculated thermodynamic Grüneisen parameter is 0.27: This small value is a consequence of the relatively low thermal expansion of beryl at 300 K (3.26 × 10−6/K, Morosin 1972). Our overall average mode gammas for the Raman bands in Table 1 is −0.05. This discrepancy with the thermodynamic value is certainly associated with our sampling of the Raman modes of beryl being highly biased by a single, low-frequency soft mode with a highly negative Grüneisen parameter.
High-pressure luminescence
In order to probe the effect of polymorphism on the luminescence spectrum, emerald was probed at high pressures through the transition observed using Raman spectroscopy. Representative high-pressure luminescence spectra on both compression and decompression are shown in Fig. 7, and the pressure shifts of the Cr3+ and V2+ emission lines are plotted in Fig. 8. The assignments and pressure shifts of the Cr3+ and V2+ emission bands are given in Table 4. The primary and secondary Cr3+ R-lines shift smoothly and to lower energy under compression up to ~13.7 GPa where the primary lines split. Above ~13.7 GPa, the two components of the R1 and R2 bands have different pressure shifts (Table 4) which, in combination with their splitting, suggests that a phase transition has occurred. The splitting is clearly manifested in the R1 band (Fig. 9), while two overlapping (but deconvolvable) components are present in the R2 band. Hysteresis of these splittings is not observed on decompression: They are fully reversible, which indicates that this transition is second order in nature. We note that this split occurs 1.7 GPa higher than the pressure determined by extrapolating polynomial extrapolation of the Raman E 1g mode. This is not surprising, since the extrapolation of this mode incorporated data only up to ~4.0 GPa, and the shift of this mode is thus less negative as the transition is approached relative to a polynomial extrapolation. Hence, our luminescence data provide an accurate constraint on the pressure that the high-pressure second-order transition occurs, and our Raman data confirm that this high-pressure transition is indeed a soft-mode transition. The pressure shift we observe up to 13.7 GPa of the primary R1 line is similar to that of the previous studies of Canny et al. (1987) and Sánchez-Alejo et al. (2011). Canny et al. (1987) report that the pressure shift of R1 in emerald of unknown origin was −10.5 (±0.5) cm−1/GPa from 0.4 to 8.0 GPa. The pressure shift of the R1 band reported by Sánchez-Alejo et al. (2011) in a natural emerald sample up to ~5 GPa was −11.3 (±0.3) cm−1/GPa. The pressure shift of our natural emerald R1 emission band is −10.7 (±0.2) cm−1/GPa, in good agreement with prior studies.

High-pressure luminescence spectra of emerald under 532-nm excitation a compression and b decompression. Notably, the broadband 4T2 emission from Cr3+ and V2+ decreases in intensity rapidly under compression

Pressure shift of emerald R-lines and 4T2 bands of a Cr3+ and b V2+. Primary and secondary R-lines are labeled with a p and s, respectively. Vertical dashed line indicates the high-pressure phase transition. Closed symbols are compression, open symbols are decompression, and error bars are smaller than the symbols

Detail of the Cr3+ R-line region showing the splitting that occurs in the high-pressure phase of emerald on a compression and b decompression
The pressure shift of the V2+ R-line in emerald is −6.5 (±0.3) cm−1/GPa. There are few previous studies of the pressure shift of V2+ R-lines in other compounds. Chopelas and Nicol (1982) report that the pressure shift of the V2+ R1 in MgO:V2+ is ~−12 cm−1/GPa, and Jovanic et al. (2010) report that the pressure shift of V2+ R1 in LaMgAl11O19:V2+ is essentially 0 cm−1/GPa. In the case of MgO, the pressure shift of V2+ is roughly two times larger than that of Cr3+ in MgO (Chopelas and Nicol 1982). The opposite seems to be the case for V2+ in emerald (our observed V2+ R1 pressure shift is about half of the Cr3+ R1 pressure shift in emerald). One key difference between the MgO study and our study is that we are characterizing the pressure shift of Cr3+ and V2+ simultaneously in the same material under compression. The unexpected smaller shift of the R-line of the larger V2+ ion may be associated with the effects of local charge balance on the environment surrounding this non-isovalent substitution: Two V2+ substitutions are plausibly accompanied by an oxygen vacancy which, if the vacancy is local to the vanadium ion, may induce a markedly different local compressibility.
The 4T2 transitions of Cr3+ and V2+ shift to higher energy up to ~8 GPa where their pressure shifts decrease, shifting only slightly to lower energy up to the maximum pressure of this study (Fig. 8a, b). The intensities of these transitions also markedly decrease under compression. The shift to higher energy and the decrease in intensity indicate that the crystal field strength changes from an intermediate strength field to a stronger field under compression to ~16.4 GPa. The shift of the 4T2 emission to higher energies and the 2E to lower energies under compression has been reported in materials doped with d 3 ions (Jovanić et al. 2010). The change in crystal field strength appears to occur continuously and across the pressure range to ~10 GPa in emerald.
The 4T2 transition corresponds to the difference in energy between the split d orbitals of the chromium ion and is represented as 10Dq. If we assume, in accord with an octahedral crystal field point-charge model, that the energy of the 4T2 transition scales as R −5Cr–O (Zheng 1995; Bray 2001), then we derive a compressibility of 6.28 × 10−4 GPa−1 for the Cr3+ bond lengths. This result is much smaller than the calculated bond compressibility of 1.96 × 10−3 GPa−1 for the Al sites in beryl using the high-pressure single-crystal results of Hazen et al. (1986) (e.g., Mei et al. 2016). Since X-ray diffraction refines an average structure, the beryl sample (synthetic emerald containing 1.3 % Cr) measured by Hazen et al. (1986) involves an average of the Al–O and Cr–O distances. Recently, Mei et al. (2016) derived covalency reduction parameters from ambient-pressure spectra of emerald and combined these with the R-line shifts of Sánchez-Alejo et al. (2011) to 5 GPa, and generated a compressibility of the Cr–O bond that was between 9 and 19 % less than that of the results of Hazen et al. (1986).
At the local scale, Gaudry et al. (2007) find that the Cr–O bond lengths in emerald are almost 4 % longer than the Al–O bond lengths and that the structural compensation for this increased bond length takes place primarily in the first coordination shell surrounding the Cr: Hence, the compaction undergone locally by the Cr ion almost certainly does not reflect the compression of the Al site within the bulk lattice. Indeed, Gaudry et al. (2007) note that the R−5 point-charge model fails to explain the magnitude of 10Dq in emerald: Our results imply that this model also, unsurprisingly, cannot explain the pressure dependence of 10Dq either. The notable disagreement between bond compressibilities derived from the combination of the point-charge model and spectroscopic pressure dependencies of Cr ion transitions and X-ray diffraction characterizations of the pressure dependence of the Al–O bond are thus not surprising and indicate that complex local bonding effects associated with Cr3+ ion substitutions may be present in beryl (e.g., Zheng 1995). Additionally, the close approach of the 4T2 and 2E states may give rise to resonance or anti-resonance effects between the levels (e.g., Sanz-Ortiz et al. 2010): Such effects, which we do not examine in detail in this study, may produce the markedly nonlinear shift of the 4T2 state in Fig. 8a. Thus, we do not present a point-charge calculation of compressibility using the pressure shift of the 4T2 emission of V2+, since it is not isovalent with Al3+ and hence we expect simple crystal field scaling rules to break down even more severely than for an isovalent substitution. In addition to local relaxation, the roles of site distortion from an octahedral environment, pressure-induced shifts in site geometry, potential complex deformation mechanisms, and spin–orbit coupling and possible admixture/resonance effects between the 4T2 and 2E levels further render such octahedral point-charge-based scaling results suspect for emerald. Nevertheless, in contrast to the modestly differing pressure shifts of the 2E R1-line emissions between vanadium and chromium, the pressure shift of the V2+ 4T2 emission is more than twice the pressure shift of the Cr3+ 4T2 emission in emerald, suggesting that sites with V2+ may be more compressible than Cr3+ sites in emerald.
The intensities of the Cr3+ and V2+ 2E transitions increase under compression (Fig. 10a) relative to the intensities of the Cr3+ and V2+ 4T2 transitions (Fig. 10b). Under compression, the energy of the 4T2 transition shifts to higher energy and moves closer in energy to the 2E transition. Indeed, by ~10 GPa, the 4T2 emission of Cr3+ lies within 5 nm of the primary R1 band. Hence, mixing between these two transitions is enhanced under compression. This increase in mixing and near-energetic equivalence likely contributes to the increase in intensity of the 2E transition under compression. It is also possible that the increase in relative intensity could be caused by the pressure shift of an absorption band relative to the wavelength of excitation: Unfortunately, no optical absorption measurements of emerald have been taken at high pressures, although significant shifts in emerald’s crystal field absorptions are observed as a function of temperature (Parkin and Burns 1980; Taran et al. 1994). Above ~10 GPa, the relative intensity of the 2E transition begins to decrease and the intensity of the 4T2 increases slightly up to the maximum pressure of this study. This observation is unusual and could be due to (1) a decrease in mixing between the 4T2 and 2E; (2) an unconstrained shift of an absorption band above 10 GPa; or (3) a significant change in the interaction between the two states across the high-pressure phase transition. By analogy with Gd3Sc2Ga3O12:Cr3+ (Hoemmerich and Bray 1995), there could be a tendency toward anti-crossing behavior of the 4T2 and 2E states in emerald under compression which starts to prevent mixing of the 4T2 and 2E near 10 GPa. With respect to pressure shifts of Cr3+ absorption bands, a shift to shorter wavelength of ~100 nm under compression to ~13.5 GPa has been reported in kosmochlor (NaCrSi2O6) (Taran et al. 2011). If such a pressure shift is present in emerald, the shape of the emerald absorption spectrum (Taran et al. 1994) is such that under compression the absorption bands of emerald could shift so that 532-nm excitation is absorbed less by the low-energy tail of the 4T1 absorption and more by the high-energy side of the 4T2 absorption. Lastly, the absorption spectrum of the high-pressure phase may be significantly different than that of the low-pressure phase. However, we do not anticipate that two Cr sites that have R1 emission lines separated by only 33.5 cm−1 (at 16.4 GPa) would generate a dramatically different absorption spectrum from the low-pressure phase.

Pressure dependence of Cr3+ and V2+ a R1 emission and b 4T2 emission. R1 intensity is normalized to the 4T2 intensity and the 4T2 intensity is normalized to the R1 intensity. Closed symbols are compression, open symbols are decompression, and error bars are smaller than the symbols
R-line splitting and FWHM
The R-line splitting is viewed as a primary manifestation of the distortion of the Al site, with the larger the splitting, the more distorted the Al site from an ideal octahedron. For example, ruby has a close-to-ideal Al site (with a modest trigonal distortion) and has an R-line splitting of ~29 cm−1 (Syassen 2008), while zoisite (Ca2Al3Si3O12(OH)) has a highly distorted Al site and has an R-line splitting of ~350 cm−1 (Koziarska et al. 1994). Emerald has a mildly distorted Al site with an R-line splitting of ~65 cm−1, and under compression to ~6.0 GPa the R-line splitting decreases by ~5 cm−1, indicating that the Al site becomes less distorted (Fig. 11a). Indeed, high-pressure single-crystal structural refinements confirm that the Al sites become mildly less distorted up to ~5.7 GPa (Hazen et al. 1986). Above ~6.0 GPa the Cr3+ R-line splitting rapidly increases up to ~13.7 GPa, suggesting that the Al sites become more distorted in this pressure interval as the high-pressure phase transition is approached. The R-line splitting of the secondary Cr3+ R-lines shows similar behavior under compression, but the data are more scattered because the second component of the R2 line is difficult to deconvolve. Indeed, the secondary R2 peak becomes impossible to resolve between ~6.5 and 13.7 GPa. Also, only the R1 component of the V2+ emission lines could be reliably fit across the pressure range of this study, and so the V2+ R-line splitting as a function of pressure was not characterized.

Pressure dependence up to ~16.4 GPa of the primary Cr3+ R-line components a R-line separation and b FWHM. Circles (green) are R1 and squares (black) are R2. Vertical dashed line indicates the high-pressure phase transition. Closed symbols are compression and open symbols are decompression
After the phase transition near 13.7 GPa, a second component of the Cr3+ R2 band again becomes resolvable, since the two components of each of the R-lines have different pressure shifts in the high-pressure phase. The pressure dependence of the Cr3+ R-line splitting changes sign and decreases above the 13.7 GPa transition (Fig. 11a). This marked change in splitting indicates that the Al sites in the high-pressure phase become less distorted under compression to the maximum pressure of this study. These variations in R-line separations are largely reversible on decompression, with the R-lines being perhaps slightly more closely separated between 13.7 and ~10 GPa on decompression relative to on compression (Fig. 6a): We have no simple explanation for this difference.
The FWHMs of the primary and secondary Cr3+ R1 bands as a function of pressure are shown in Fig. 11b. The FWHM of the R1 band decreases up to ~6.0 GPa, and from ~6.0 to the ~13.7 GPa phase transition, the FWHM of both the R1 and R2 bands increases. Above the transition, the FWHMs decrease up to the maximum pressure of 16.4 GPa. On decompression, the FWHMs of the R-lines generally follow the same trend as they did during compression, although the scatter is substantially greater, possibly due to the effects of residual strain following decompression. The Cr3+ R-line FWHM begins to increase at ~6.0 GPa, well below the pressure of the glass transition in methanol/ethanol mixtures (Piermarini et al. 1973; Klotz et al. 2009), and thus the broadening cannot be attributed to non-hydrostaticity. This trend of decreased splitting and FWHM above 13.7 GPa of the R-lines is thus robust: As such, the decreases in these parameters following the transition reflect a fundamental shift in the response to pressure of the Al/Cr site within the high-pressure phase.
The pressure dependence of the FWHM of the V2+ R1 line is challenging to resolve from our peak fits, and this peak may include difficult-to-deconvolve components of the 4T2 emission bands of both Cr3+ and V2+. The room-pressure and room-temperature FWHM of the V2+ R1 that we determine from our deconvolution is ~200 cm−1. This is extremely wide for a V2+ R1 emission line (e.g., the FWHM of the V2+ R1 line in MgO is ~7 cm−1 at room pressure and temperature, Imbusch et al. 1964) and may reflect an association of this band with a vacancy, or a structurally distorted environment. The pressure dependence of the linewidth of R-lines can be modeled using a Debye-based model (Kottke and Williams 1983), which is modified from the formulation for the temperature dependence of the R-line widths (Imbusch et al. 1964). Since vibronic modes and neighbor lines could not be resolved across the pressure range of these measurements (due to the broad 4T2 emission bands of both Cr3+ and V2+), we do not know the rates at which vibronic modes shift, and whether they are anticipated to interact more or less strongly with the R-lines at high pressures. Moreover, our high-pressure Raman experiments provide the only experimental constraints on beryl’s phonon spectrum under pressure. The vibronic modes that primarily couple with the R1-lines to generate their width may initially shift at rates faster than the R-lines, which would generate the initial decrease in R-line widths under compression to ~6.0 GPa. The decrease in peak widths above the transition may reflect either a structural effect (a more symmetric environment of the multiple Cr/Al sites within a different phase, and hence a smaller intrinsic peak width), or a shift in phonon coupling in the high-pressure phase, or a complementary combination of these two effects.
High-pressure phase transition
Dramatic changes in the Raman spectra at ~14 GPa, a negative pressure shift of the E 1g mode, and splitting of the Cr3+ R-lines at ~13.7 GPa all indicate that beryl undergoes a phase transition to beryl-II near 14 GPa. The absence of hysteresis in both the Raman and luminescence spectra suggests that this transition is second order in nature. Thus, both types of spectroscopic data are in accord with the predicted transition at 14 GPa proposed by Prencipe et al. (2011). Our Raman data also confirm that this transition is driven by a soft mode (Fig. 6a). Our spectroscopic data allow us to make some general comments on the high-pressure deformation of beryl up to and above the transition to beryl-II. Indeed, the detailed changes in the Si6O18 ring and Al/Be polyhedra under compression were only characterized up to 5.7 GPa in single-crystal diffraction measurements by Hazen et al. (1986).
Our Raman and luminescence data indicate that the crystal structure of beryl accommodates initial compression with little or no distortion of the Si6O18 rings. Indeed, our high-pressure Raman and luminescence data (the pressure shift of emission lines, R-line splitting, and FWHM) suggest that the compaction mechanism of beryl involves primarily compression of the Al and Be sites up to ~6.0 GPa. Above ~6.0 GPa, the deformation mechanism may include compaction of the Si6O18 rings, based on the changes observed in R-line splitting and FWHM of the Cr3+ R-lines. However, the deformation of the Si6O18 rings above ~6.0 GPa is still likely minimal since no obvious changes in the Raman spectra are observed until above ~12 GPa. High-pressure single-crystal structures of beryl reported by Hazen et al. (1986) show that the components of the rigid Si6O18 rings do not significantly distort under compression up to 5.7 GPa, as indicated by the quadratic elongations (Robinson et al. 1971) of the Si tetrahedra. This is in contrast to the quadratic elongation of the Al/Be sites reported by Hazen et al. (1986). The increased distortion of the Al/Cr sites manifested by the shift near 6 GPa in the splitting and FWHM luminescence data indicates that the onset of larger degrees of distortion of the Al polyhedra may be associated with the initiation of deformation of the tetrahedra of the Si6O18 rings (Fig. 1). The deformation of the rings above ~6.0 GPa likely includes mild distortions of the Si tetrahedra (changes in bond angles or movement of the Si atom), but these changes do not generate a change in symmetry. Our luminescence data thus suggest that a change in the compressional mechanism of emerald occurs at ~6.0 GPa, with the Al sites becoming increasingly distorted up to 13.7 GPa. Notably, quantum mechanical calculations on the compression of beryl up to 28.4 GPa show that the O2 atoms (which link the Al and Be sites) move away from the Si atoms and closer to the Al and Be atoms, while the O1 atoms, located on the inside of the Si6O18 rings, move in the ab plane toward the center of the Si6O18 rings (Fig. 1; Prencipe and Nestola 2005). Notably, the existence of the soft-mode transition was unknown at the time of these calculations, and the calculations above 14 GPa were done using the symmetry of the low-pressure phase.
At 13.7 GPa, the Cr3+ R1 band becomes asymmetric (Fig. 9), and a change in the pressure dependence of the V2+ R-line is observed (Fig. 8b). On increasing pressure above ~13.7 GPa, the Cr3+ R1 line clearly splits into two components, and the Cr3+ R2 line also becomes asymmetric. Dramatic changes in the trends of the FWHM as well as a splitting of the Cr3+ R-lines also occur at ~13.7 GPa (Fig. 11). Our Raman spectra show discontinuous shifts of all of the observed Raman modes as well as a dramatic decrease in intensity and FWHM of all observed Raman modes. Clearly, a phase transition that shifts the geometry and response to pressure of the Cr/Al site has taken place, as well as a change in the local bonding environment of the Si tetrahedra. This transition appears to be second order in nature, since no significant hysteresis of either the luminescent bands or Raman spectra is observed on decompression. The different pressure shifts of the two components of the Cr3+ R-lines in the high-pressure phase strongly suggest that there are two crystallographically distinct Al sites with different responses to pressure in the high-pressure phase. These observations of a phase transition near 13.7 GPa are in agreement with the proposed soft-mode-associated high-pressure transition predicted by Prencipe et al. (2011). Our Raman data, with a strongly negative shift of a low-frequency band, are in full accord with this predicted soft-mode transition. The similarity of the decreases in R-line splitting and FWHM between 0 and 3 GPa, and above 13.7 GPa, suggests that the initial deformation mechanism of the Al sites in the high-pressure phase of emerald may be generally similar to that from room pressure to ~3.0 GPa. Hence, following the deformation of the Al sites above ~6 GPa accompanied by ring deformation, the combination of deformation of the components of the rings, and the evolution of two Al sites may allow for deformation to primarily be accommodated by the Al and Be sites. This deformation results in the Al sites becoming more regular up to the maximum pressure of this study, as shown by the decreases in R-line separation and FWHM above ~13.7 GPa.
The hybrid density functional theory (DFT) calculations of Prencipe et al. (2011) predict that a transition near 14 GPa from hexagonal to triclinic symmetry involves splitting the single Si site in hexagonal beryl to three Si sites in the high-pressure triclinic beryl. Our high-pressure Raman spectra do not evolve additional peaks in the high-pressure beryl-II spectra, although pervasive peak broadening is observed (Fig. 5). Thus, the bonding environment of the three Si sites is likely similar in beryl-II.
The ring silicate cordierite ((Mg,Fe)2Al4Si5O18) shows potentially analogous behavior under compression: Recent high-pressure single-crystal structure determinations show that extensive deformation of cordierite’s silicate rings occurs when orthorhombic cordierite transforms to triclinic cordierite at ~7.5 GPa (Miletich et al. 2014a; Finkelstein et al. 2015). Rings become more oval shaped, and the Si sites not only split into multiple sites, but coordination changes in the octahedral and tetrahedral sites between the silicate rings also occur. The changes occurring in emerald at ~13.7 GPa are not as dramatic as those that occur in cordierite. No changes in coordination of the Al sites are observed: These would be expected to produce dramatic changes in the character of the Cr3+ luminescence. Changes in the coordination of the Si sites are also not occurring in beryl-II. The notably different high-pressure behavior of these two ring silicates is thus likely related to the octahedrally coordinated Al in beryl being more stable under compression than the tetrahedrally coordinated Al in cordierite. Hence, the Si6O18 ring of beryl is stable to higher pressures than the Si6O18 ring of cordierite, and changes in coordination of Al, Si, or Be are not likely up to ~16.4 GPa. Therefore, H2O and other volatiles would remain stable inside the channels of beryl to higher pressures than in cordierite. It is also is clear that disordering does not take place in emerald to 16.4 GPa: In silicate glasses, R-lines are broadened, merged into one emission peak, and shifted in wavelength, and the broad 4T2 emission dominates the spectrum (Yamaga and Henderson 1991). Even in these disordered environments, Cr3+ strongly prefers octahedral coordination due to its high stabilization energy (Shinn and Tesar 1992). Thus, even in these extreme examples, Cr3+ emission occurs from a site that is sixfold coordinated, and the emission spectrum still has the characteristic features of Cr3+ in octahedral coordination. Hence, changes in coordination of the Al/Cr site and the Si site do not occur across the observed ~13.7 GPa phase transition in emerald.
Previous high-pressure powder diffraction
Two high-pressure powder diffraction studies have reached pressures in excess of ~14 GPa, and no high-pressure phase transitions have been reported (Qin et al. 2008; Fan et al. 2015). It is possible that, since the thermal expansion of different beryl compositions can depend strongly on their impurity content (Morosin 1972; Schlenker et al. 1977; Hochella and Brown 1986), minor compositional differences may play a role in the high-pressure polymorphism of this material. However, the effect of a few thousand ppm of Cr and V would be far more likely to simply offset a phase transition to modestly higher (or lower) pressures. A more likely possibility is that the transition in beryl involves twinning (e.g., Prencipe et al. 2011) or a transition to a mildly modulated structure. A drop from hexagonal to triclinic symmetry would likely result in extensive twinning, and often in powder diffraction patterns this twinning may appear to resemble the higher symmetry lattice. Hence, without the correct twin law, these patterns could be refined within a hexagonal crystal system. Moreover, the volume and lattice parameters as a function of pressure reported by Qin et al. (2008) and Fan et al. (2015) do not agree with one another (see supplementary Figures S1 and S2).
The refinement of incommensurately modulated structures from powder diffraction data is difficult, and usually single-crystal data are required to accurately determine the full symmetry of modulated phases. Also, overexposure techniques would need to be employed in order to properly resolve the intensities of satellite peaks. Satellite peaks can be missed if the assumption is made that no change from hexagonal symmetry occurs across the pressure range of the experiment. Limitations in angular access of typical diamond anvil cells may render detection of a transition to a modulated structure difficult to detect using standard Le Bail refinement techniques. Such modulated structures are precedented in ring silicates: Cordierite undergoes a transition from orthorhombic to hexagonal at high temperature (1450 °C) via a continuous mechanism that involves a metastable short-range-ordered modulated structure (Putnis and Bish 1983). Moreover, the E 1g mode at 138 cm−1 involves a dynamic wave-like distortion of the entire structure of beryl [again see animations on the CRYSTAL Web site1]. It is likely that the soft-mode transition freezes in this distortion, which could lead to twinning or a modulated structure.
Conclusions
High-pressure Raman and luminescence spectra have been collected for beryl and emerald up to a maximum pressure of ~16.4 GPa. Using both techniques, we have characterized the high-pressure behavior of the Al, Be, and Si sites within beryl under compression and decompression. Our high-pressure Raman spectra show that an E 1g symmetry mode (ring flexure) located at 138 cm−1 under ambient conditions shifts negatively under compression. Both our observations and an extrapolation of this band’s mode shift agree well with the pressure shift of this soft mode calculated by Prencipe et al. (2011). Hence, our Raman data provide the first experimental evidence of the proposed soft-mode transition, and our luminescence data confirm that a modest structural change occurs near 14 GPa. Both sharp line and broadband Cr3+ and V2+ luminescence are observed and characterized at both ambient- and high-pressure conditions. The pressure shifts of Cr3+ and V2+ differ under compression, and this difference is likely related to the different valence states and local charge compensation associated with these two transition metal elements. The Al sites likely undergo increasing distortion above ~6.0 GPa, as manifested by shifts in R-line splitting and FWHM near this pressure. Splitting of luminescent bands and changes in the sign of trends in FWHM are associated with a second-order phase transition at ~13.7 GPa. Our Raman and luminescence results are in good agreement with the second-order, soft-mode phase transition from P6/mcc to P\(\bar{1}\) predicted by ab initio calculations near ~14 GPa by Prencipe et al. (2011), and observed by M. Merlini in high-pressure single-crystal measurements (personal communication): We designate the new phase beryl-II. We observed the transition under non-hydrostatic conditions, and it is possible that the pressure that this transition occurs under hydrostatic conditions may modestly differ. However, the soft-mode mechanism of the transition appears robust, since the soft mode is observed to shift negatively under hydrostatic conditions. Moreover, the R-line separation and FWHM in the luminescence spectrum of beryl do not dramatically increase above 10.5 GPa, suggesting that pressure gradients are small. The chemical composition (and particularly the level of Cr substitution) is known to change the high-temperature behavior of beryl, but it does not appear that the level of Cr substitution dramatically changes the high-pressure behavior of beryl, since the transition was observed at similar pressures in emerald (in luminescence) and beryl (in Raman spectra). Twinning likely occurs across this transition, and the transition might also be to a mildly modulated structure. The high-pressure phase of beryl might have interesting lasing properties, since Cr3+ is in a strong crystal field in the high-pressure phase. The intense Cr3+ R1 emission line has two components separated by 1.60 nm (33.5 cm−1) at ~16.4 GPa, and at higher pressures, these bands likely become increasingly separated. Hence, these emission lines could be selectively excited across a modest wavelength range.
References
Aurisicchio C, Fioravanti G, Grubessi O, Zanazzi PF (1988) Reappraisal of the crystal chemistry of beryl. Am Miner 73:826–837
Bray KL (2001) High pressure probes of electronic structure and luminescence properties of transition metal and lanthanide systems. In: Yersin H (ed) Transition metal and rare earth compounds. Springer, Berlin, Heidelberg, pp 1–94
Buchert J, Katz A, Alfano RR (1983) Laser action in emerald. IEE J Quantum Electron 19:1477–1478
Burns R (1993) Mineralogical applications of crystal field theory, 2nd edn. Cambridge University Press, Cambridge
Canny B, Chervin JD, Curie D, Gonzalez J, Berry D, Ho SA (1987) High pressure properties of some laser materials. In: Di Bartolo B (ed) Spectroscopy of solid-state laser-type materials. Springer, US, pp 431–449
Chopelas A, Nicol M (1982) Pressure dependence to 100 kilobars of the phonons of MgO at 90 and 295 K. J Geophys Res 87:8591–8597
Fan D, Xu J, Kuang Y, Li X, Li Y, Xie H (2015) Compressibility and equation of state of beryl (Be3Al2Si6O18) by using a diamond anvil cell and in situ synchrotron X-ray diffraction. Phys Chem Miner 42:529–539
Finkelstein GJ, Dera PK, Duffy TS (2015) High-pressure phases of cordierite from single-crystal X-ray diffraction to 15 GPa. Am Miner 100:1821–1833
Fridrichová J, Bačík P, Bizovská V, Libowitzky E, Škoda R, Uher P, Ozdin D, Števko M (2016) Spectroscopic and bond-topological investigation of interstitial volatiles in beryl from Slovakia. Phys Chem Miner 43:419–437
Fujishiro I, Piermarini GJ, Block S, Munro RG (1982) Viscosities and glass transition pressures in the methanol–ethanol–water system. In: High pressure research in science and industry: proceedings of the 8th AIRAPT conference, vol 2, pp 608–611
Gaft M, Reisfeld R, Panczer G (2005) Modern luminescence spectroscopy of minerals and materials. Springer, Berlin
Gaudry E, Cabaret D, Brouder C, Letard I, Rogalev A, Wilhlem F, Jaouen N, Sainctavit P (2007) Relaxations around the substitutional chromium site in emerald: X-ray absorption experiments and density functional calculations. Phys Rev B 76:094110
Gibbs G, Breck D, Meagher E (1968) Structural refinement of hydrous and anhydrous synthetic beryl and emerald. Lithos 1:275–285
Hagemann H, Lucken A, Bill H, Gysler-Sanz J, Stalder HA (1990) Polarized Raman spectra of beryl and bazzite. Phys Chem Miner 17:395–401
Hazen RM, Au AY, Finger W (1986) High-pressure crystal chemistry of beryl (Be3Al2Si6O18) and euclase (BeAlSiO4OH). Am Miner 71:977–984
Hemingway BS, Barton MD, Robie RA, Haselton HT (1986) Heat capacities and thermodynamic functions for beryl, Be3Al2Si6O18, phenakite, Be2SiO4, euclase, BeAlSiO4(OH), bertrandite Be4Si2O7(OH)2, and chrysoberyl BeAl2O4. Am Miner 71:557–568
Hochella MF, Brown GE (1986) Structural mechanisms of anomalous thermal expansion of cordierite-beryl and other framework silicates. J Am Ceram Soc 69:13–18
Hoemmerich U, Bray KL (1995) Direct observation of anticrossing behaviour in a luminescent Cr3+-doped system. Phys Rev B 51:8595–8598
Hofmeister AM, Hoering TC, Virgo D (1987) Vibrational spectroscopy of beryllium aluminosilicates: heat capacity calculations from band assignments. Phys Chem Miner 14:205–224
Imbusch GF, Yen WM, Schawlow AL, McCumber DE, Sturge M (1964) Temperature dependence of the width and position of the 2E 4A2 fluorescence lines of Cr3+ and V2+ in MgO. Phys Rev 133:A1029–A1034
Jovanić BR (1997) High pressure and fluorescence lifetime of the 2E level in MgO:Cr3+. Phys Scr 56:477–479
Jovanić BR (2000) Effect of high pressure on fluorescence lifetime and position for R1 line in synthetic spinel MgAl2O4:Cr3+. Mater Sci Forum 352:247–250
Jovanić BR, Viana B, Radenković B, Despotović M, Panić B (2010) High-pressure optical studies of LMA: V2+. Mater Chem Phys 124:109–112
Kisliuk P, Moore CA (1967) Radiation from the 4T2 State of Cr3+ in ruby and emerald. Phys Rev 160:307–312
Klotz S, Chervin J-C, Munsch P, Le Marchand G (2009) Hydrostatic limits of 11 pressure transmitting media. J Phys D Appl Phys 42:075413
Kottke T, Williams F (1983) Pressure dependence of the alexandrite emission spectrum. Phys Rev B 28:1923–1927
Koziarska B, Godlewski M, Suchocki A, Czaja M, Mazurak Z (1994) Optical properties of zoisite. Phys Rev B 50:297–300
Lai ST (1987) Highly efficient emerald laser. J Opt Soc Am B 4:1286–1290
Likhacheva AY, Goryainov SV, Krylov AS, Bul’bak TA, Prasad P (2012) Raman spectroscopy of natural cordierite at high water pressure up to 5 GPa. J Raman Spectrosc 43:559–563
Mao HK, Xu JA, Bell PM (1986) Calibration of the ruby pressure gauge to 800 kbar under quasi-hydrostatic conditions. J Geophys Res 91:4673–4676
Mei Y, Peng RM, Chen BW, Zheng WC (2016) Local compressibilities of Cr3+ octahedral clusters in the natural and synthetic emerald crystals. Optik 127:5152–5154
Milberg ME, Blair HD (1977) Thermal expansion of cordierite. J Am Ceram Soc 60:372–373
Miletich R, Gatta GD, Willi T, Mirwald PW, Lotti P, Merlini M (2014a) Cordierite under hydrostatic compression: anomalous elastic behavior as a precursor for a pressure-induced phase transition. Am Mineral 99:479–493
Miletich R, Scheidl KS, Schmitt M, Moissl AP, Pippinger T, Gatta GD, Schuster B, Trautmann C (2014b) Static elasticity of cordierite I: effect of heavy ion irradiation on the compressibility of hydrous cordierite. Phys Chem Miner 41:579–591
Morosin B (1972) Structure and thermal expansion of beryl. Acta Crystallogr Sect B Struct Crystallogr Cryst Chem 28:1899–1903
O’Bannon E, Williams Q (2016) A Cr3+ luminescence study of spodumene at high pressures: effects of site geometry, a phase transition and a level-crossing. Am Miner 101:1406–1413
Ohkura H, Hashimoto H, Mori Y, Chiba Y, Isotani S (1987) The luminescence and ESR of a synthetic emerald and the natural ones mined from Santa Terezinha in Brazil. Jpn J Appl Phys 26:1422–1428
Ollier N, Fuchs Y, Cavani O, Horn AH, Rossano S (2015) Influence of impurities on Cr3+ luminescence properties in Brazilian emerald and alexandrite. Eur J Miner 27:783–792
Parkin KM, Burns RG (1980) High temperature crystal field spectra of transition metal-bearing minerals: relevance to remote-sensed spectra of planetary surfaces. In: Lunar and planetary science conference proceedings, vol 11, pp 731–755
Piermarini GJ, Block S, Barnett JD (1973) Hydrostatic limits in liquids and solids to 100 kbar. J Appl Phys 44:5377–5382
Piermarini GJ, Block S, Barnett JD, Forman RA (1975) Calibration of the pressure dependence of the R1 ruby fluorescence line to 195 kbar. J Appl Phys 46:2774
Prencipe M, Nestola F (2005) Quantum-mechanical modeling of minerals at high pressures. The role of the Hamiltonian in a case study: the beryl (Al4Be6Si12O36). Phys Chem Miner 32:471–479
Prencipe M, Nestola F (2007) Minerals at high pressure. Mechanics of compression from quantum mechanical calculations in a case study: the beryl (Al4Be6Si12O36). Phys Chem Miner 34:37–52
Prencipe M, Noel Y, Civalleri B, Roetti C, Dovesi R (2006) Quantum-mechanical calculation of the vibrational spectrum of beryl (Al4Be6Si12O36) at the Γ point. Phys Chem Miner 33:519–532
Prencipe M, Scanavino I, Nestola F, Merlini M, Civalleri B, Bruno M, Dovesi R (2011) High-pressure thermo-elastic properties of beryl (Al4Be6Si12O36) from ab initio calculations, and observations about the source of thermal expansion. Phys Chem Miner 38:223–239
Putnis A, Bish DL (1983) The mechanism and kinetics of Al, Si ordering in Mg-cordierite. Am Miner 68:60–65
Qin S, Liu J, Li H, Zhu X, Li X (2008) In-situ high-pressure X-ray diffraction of natural beryl. Chin J High Press Phys 22:1–5
Quarles GJ, Suchocki A, Powell RC, Lai S (1988) Optical spectroscopy and four-wave mixing in emerald. Phys Rev B 38:9996–10006
Robinson K, Gibbs GV, Ribbe PH (1971) Quadratic elongation: a quantitative measure of distortion in coordination polyhedra. Science 172:567–570
Sánchez-Alejo MA, Hernández-Alcántara JM, Jiménez C, Calderón T, García EC (2011) Optical spectroscopy and high pressure on emeralds: synthetic and natural. In: Proceedings of SPIE, the international society for optical engineering, vol 8011, pp 1–6
Sanz-Ortiz MN, Rodriguez F, Hernandez I, Valiente R, Kuck S (2010) Origin of the 2E–4T2 Fano-type resonance in Cr3+-doped LiCaAlF6: pressure-induced excited-state crossover. Phys Rev B 81:045114
Schlenker JL, Gibbs GV, Hill EG, Crews SS, Myers RH (1977) Thermal expansion coefficients for indialite, emerald, and beryl. Phys Chem Miner 1:243–255
Schmetzer K, Eysel HH (1974) Absorptions- and emissonsspektrum von V2+/V3+ in beryllen. Zeitschrift für Naturforsch A 29:1458–1460
Shen Y, Riedener T, Bray KL (2000) Effect of pressure and temperature on energy transfer between Cr3+ and Tm3+ in Y3Al5O12. Phys Rev B 61:11460–11471
Sherriff B, Grundy D, Hartman JS, Hawthorne F, Cerny P (1991) The incorporation of alkalis in beryl: multi-nuclear MAS NMR and crystal-structure study. Can Miner 29:271–285
Shinn MD, Tesar A (1992) Observation of temperature-dependent Cr3+–Nd3+ sensitization in fluoride glasses. J Lumin 51:189–195
Skvortsova V, Mironova-Ulmane N, Trinkler L, Merkulov V (2015) Optical properties of natural and synthetic beryl crystals. In: IOP conference series: materials science and engineering, vol 77, p 012034
Snytnikov VN, Stoyanovskii VO, Larina TV, Krivoruchko OP, Ushakov VA, Parmon VN (2008) Laser-induced luminescence of model Fe/Al2O3 and Cr/Al2O3 catalysts. Kinet Catal 49:291–298
Sugano S, Tanabe Y (1958) Absorption spectra of Cr3+ in Al2O3 part A. Theoretical studies of the absorption bands and lines. J Phys Soc Jpn 13:880–889
Syassen K (2008) Ruby under pressure. High Press Res 28:75–126
Taran MN, Langer K, Platonov AN, Indutny VV (1994) Optical absorption investigation of Cr3+ ion-bearing minerals in the temperature range 77–797 K. Phys Chem Miner 21:360–372
Taran MN, Ohashi H, Langer K, Vishnevskyy AA (2011) High-pressure electronic absorption spectroscopy of natural and synthetic Cr3+-bearing clinopyroxenes. Phys Chem Miner 38:345–356
Wamsley P, Bray K (1994) The effect of pressure on the luminescence of Cr3+:YAG. J Lumin 59:11–17
Williams Q, Jeanloz R (1985) Pressure shift of Cr3+-ion-pair emission lines in ruby. Phys Rev B 31:7449–7451
Wood DL, Nassau K (1967) Infrared spectra of foreign molecules in beryl. J Chem Phys 47:2220–2228
Xu J, Kuang Y, Zhang B, Liu Y, Fan D, Li X, Xie H (2016) Thermal equation of state of natural tourmaline at high pressure and temperature. Phys Chem Miner 43:315–326
Yamaga M, Henderson B (1991) Line shape and lifetimes of Cr3+ luminescence in silicate glasses. Phys Rev B 44:4853–4861
Yoon HS, Newnham RE (1973) The elastic properties of beryl. Acta Crystallogr Sect A 29:507–509
Zheng W-C (1995) Determination of the local compressibilities for Cr3+ ions in some garnet crystals from high-pressure spectroscopy. J Phys Condens Matter 7:8351–8356
Acknowledgments
Constructive and insightful comments from M. Prencipe and an anonymous reviewer, and helpful discussions with M. Merlini and C.M. Beavers, greatly improved the quality of this manuscript. E. O’Bannon thanks Juan Diego Prieto for the emerald sample. We thank Dan Sampson for invaluable technical assistance with the Raman spectrometer and Rob Franks for help with the trace element analysis. Work was partially supported by NSF through EAR-1215745, and COMPRES, the Consortium for Materials Properties Research in Earth Sciences under NSF Cooperative Agreement EAR 11-57758. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
O’Bannon, E., Williams, Q. Beryl-II, a high-pressure phase of beryl: Raman and luminescence spectroscopy to 16.4 GPa. Phys Chem Minerals 43, 671–687 (2016). https://doi.org/10.1007/s00269-016-0837-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-016-0837-2