
Abstract
The chemical composition and the crystal structure of pezzottaite [ideal composition Cs(Be2Li)Al2Si6O18; space group: \({\it{R}} \overline{\text{3}} \) c, a = 15.9615(6) Å, c = 27.8568(9) Å] from the type locality in Ambatovita (central Madagascar) were investigated by electron microprobe analysis in wavelength dispersive mode, thermo-gravimetric analysis, Fourier-transform infrared spectroscopy, single-crystal X-ray (at 298 K) and neutron (at 2.3 K) diffraction. The average chemical formula of the sample of pezzottaite resulted Cs1,Cs2(Cs0.565Rb0.027K0.017)Σ0.600 Na1,Na2(Na0.101Ca0.024)Σ0.125Be2.078Li0.922 Al1,Al2(Mg0.002Mn0.002Fe0.003Al1.978)Σ1.985 Si1,Si2,Si3(Al0.056Si5.944)Σ6O18·0.27H2O. The (unpolarized) IR spectrum over the region 3,800–600 cm−1 was collected and a comparison with the absorption bands found in beryl carried out. In particular, two-weak absorption bands ascribable to the fundamental H2O stretching vibrations (i.e. 3,591 and 3,545 cm−1) were observed, despite the mineral being nominally anhydrous. The X-ray and neutron structure refinements showed: (a) a non-significant presence of aluminium, beryllium or lithium at the Si1, Si2 and Si3 sites, (b) the absence (at a significant level) of lithium at the octahedral Al1, Al2 and Al3 sites and (c) a partial lithium/beryllium disordering between tetrahedral Be and Li sites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pezzottaite is a rare Cs-bearing mineral with ideal composition Cs(Be2Li)Al2Si6O18 (Laurs et al. 2003; Hawthorne et al. 2004). It is considered as one of the members of the “beryl group”, along with beryl sensu scricto (Be3Al2Si6O18; Aurisicchio et al. 1988; Gatta et al. 2006), bazzite (Be3Sc2Si6O18, Armbruster et al. 1995) and stoppaniite (Be3Fe2Si6O18, Ferraris et al. 1998; Della Ventura et al. 2000).
Chemical and gemmological characteristics of pezzottaite from the Sakavalana granitic pegmatite located near Ambatovita (Madagascar), along with a geological description of the deposit, were reported by Laurs et al. (2003) and Hawthorne et al. (2004). Later, the crystal structure of pezzottaite was solved and refined by Yakubovich et al. (2009) by single-crystal X-ray diffraction, in the space group \({\it{R}} \overline{\text{3}} \) c with the unit-cell parameters a ~ 15.9 and c ~ 27.8 Å. Pezzottaite is the only member of the beryl group with a rhombohedral lattice, all the other members show a primitive lattice (space group P6/mcc) with a ~ 9.2 Å and c ~ 9.2 Å.
Yakubovich et al. (2009) provided a comparative study of pezzottaite and “vorobyevite” (“morganite”, a Cs-rich beryl) [chemical formula: (Cs0.08Na0.42)[Al2(Be2.35Li0.65)Si6O18]·0.18H2O, with the space group P6/mcc and unit-cell parameters a = 9.2102(14) Å, c = 9.2179(14) Å]. The crystal structure of pezzottaite and Cs-rich beryl are topologically identical, though not iso-symmetric. The structure consists of six-membered rings of SiO4 tetrahedra, perpendicular to the [0001] axis, linked by AlO6 octahedra and (Li, Be)O4 tetrahedra that form a three-dimensional “framework” (Fig. 1). Alkali cations (e.g. Cs, Rb, K, Na) and H2O molecules represent the so-called “extra-framework” content, which lies within the six-membered ring channels along [0001]. The alkali cations compensate the positive charge deficit due to Li+ ↔ Be2+ substitution (Černý 2002; Černý et al. 2003). In pezzottaite, the Be/Li distribution in the tetrahedral sites was described as fully ordered (leading to a lowering of the symmetry if compared with beryl), whereas in “vorobyevite” the Be/Li distribution was modelled as fully disordered (Yakubovich et al. 2009). In addition, the Cs content in pezzottaite was found being significantly higher than that in “vorobyevite” (i.e. Cs2O ~ 18 wt% in pezzottaite, Cs2O < 9 wt% in Cs-rich beryl). The pezzottaite structure was modelled as completely anhydrous (though evidence of H2O content was found up to ~2 wt%; Laurs et al. 2003; Simmons et al. 2003; Hawthorne et al. 2004; Liu and Peng 2005; Yakubovich et al. 2009), whereas “vorobyevite” was modelled with extra-framework H2O. The presence of H2O (along with Na) is common as channel content in natural beryls.

The crystal structure of pezzottaite (viewed down [0001]) and the bonding configuration of the channel content based on the structure refinement of this study
Some open questions concern the crystal chemistry of Cs-rich beryl/pezzottaite. For example, the amount of Cs (and so the Li/Be content) is still unknown at which the structure transforms from P6/mcc to \({\it{R}} \overline{\text{3}} \) c. In pezzottaite, the Li/Be distribution in the tetrahedral sites was modelled as completely ordered; however, the similar X-ray scattering factors of Li and Be do not allow an unambiguous description of the ordering. In addition, the chemical data of pezzottaite so far reported show a systematic deficit of Li (~0.1 atoms per formula unit, a.p.f.u.), coupled with an excess of Be, though (Li + Be) = 3 a.p.f.u. (Laurs et al. 2003; Simmons et al. 2003; Hawthorne et al. 2004; Yakubovich et al. 2009). If H2O is also present in pezzottaite channels, the configuration of the molecules is still unknown (Yakubovich et al. 2009).
In this light, the aim of the present study is a multi-methodological investigation of a pezzottaite from Ambatovita, central Madagascar, kindly provided by F. Pezzotta (Natural History Museum of Milan), by means of electron microprobe analysis in wavelength dispersive mode (EMPA-WDS), thermo-gravimetric analysis (TG), Fourier-transform infrared (FTIR) spectroscopy, single-crystal X-ray and neutron diffraction, in order to describe (1) the Li/Be ordering in the tetrahedral sites, and excluding the presence of Li in the octahedral sites (see Yakubovich et al. 2009 and references therein), (2) the location of the oxygen and proton sites of the H2O molecules, if present at a significant level, and of the atomic relationship via the H bonds and (3) a comparison of the IR spectrum of pezzottaite with that of beryl. Due to the different scattering lengths of Li and Be (i.e. b(Li) = −1.90 fm, b(Be) = 7.79 fm, Sears 1986), single-crystal neutron diffraction represents the most appropriate experimental technique that will allow us to answer the open questions about the crystal structure of pezzottaite. This study follows a series of our recent investigations on Cs-rich aluminosilicates in search of potential nuclear waste disposal materials (Gatta et al. 2008, 2009a, b, 2010).
Experimental methods
A gem-quality, transparent and bright pink crystal of pezzottaite (~5 cm3) was used for the present multi-methodological study. The sample comes from the type locality in Ambatovita (central Madagascar), and in particular from the Sakavalana pegmatite, located in the northern part of the famous Ampandramaika-Malakialina pegmatite district (Laurs et al. 2003). The crystal sample was cut into a few fragments in order to perform EMPA-WDS, TG, FTIR, single-crystal X-ray and neutron diffraction experiments.
Quantitative chemical analyses in wavelength dispersion of a large fragment (1 mm2) were performed using a Jeol JXA-8200 electron microprobe. The system was operated with an accelerating voltage of 15 kV, a beam current of 15 nA and a counting time of 60 s on the peaks and 30 s on the background. The following elements were measured using a series of minerals as standards: Cs (pollucite), Rb (pollucite), K (K-feldspar), Na (omphacite), Ca (wollastonite), Mg (forsterite), Mn (rhodonite), Fe (fayalite), Si (wollastonite), Al (anorthite) and Ti (ilmenite). The results were processed for matrix effects using a conventional ΦρZ routine in the Jeol suite of programmes (Armstrong 1988). The crystal was found to be chemically homogeneous. The proportional formula was calculated using the protocol of Laurs et al. (2003) (i.e. with a normalization on 18 oxygen atoms, Be and Li assumed to be equal to 2 and 1 a.p.f.u., respectively, and H2O based on the TG analysis).
Thermo-gravimetric analyses were performed with a NETZSCH STA449C thermoanalyser under a 20-mL min−1 air flux with a heating rate of 5 °C min−1, using a powdered sample (crystallite diameter 2–5 μm) previously dried at 70 °C for 4 h. The experiment was conducted up to 1,370 K. The weight loss was about 0.4 % within the T-range investigated, which is ascribable to H2O loss. A representative number of EMPA-WDS/TG analyses are listed in Table 1.
The infrared spectroscopic investigation in the mid-infrared (3,800–600 cm−1) range was carried out using a Nicolet Nexus FTIR spectrophotometer. The spectrum was collected in transmission mode by KBr compressed pellet, operating at a resolution of 4 cm−1.
Two crystals of approximately 2.8 × 2.4 × 2.0 mm3 and 0.36 × 0.31 × 0.18 mm3, free of zoning under a polarized microscope, were selected for the neutron and X-ray diffraction experiment, respectively. X-ray intensity data were collected at 298 K and up to 2θmax = 72.4° (with −21 ≤ h ≤ 21, −25 ≤ k ≤ 25 and −46 ≤ l ≤ 45, Table 2) with an Xcalibur diffractometer equipped with CCD and monochromatized Mo-Kα radiation and operated at 50 kV and 40 mA. The data collection was performed with a combination of φ and ω scans, fixing a step size of 1° and a time of 10 s/frame. A total number of 48,684 reflections were collected, giving a metrically rhombohedral lattice (here in hexagonal setting) with a = 15.9615 (6) Å, c = 27.8568(9) Å and V = 6,146.2(4) Å3 (Table 2), and the systematic absences suggested the space group \({\it{R}} \overline{\text{3}} \) c, as previously reported for pezzottaite (Yakubovich et al. 2009). The intensity data were then integrated with the computer programme CrysAlis (Agilent Technologies 2012), and corrected for Lorentz-polarization effects. An absorption correction was applied by Gaussian integration based upon the physical description of the crystal (CrysAlis, Agilent Technologies 2012). After the corrections, the discrepancy factor amongst symmetry-related reflections (Laue class: \( \overline{3} \) m) was R int = 0.0393 (Table 2).
The single-crystal neutron diffraction experiment was performed on the hot single-crystal neutron diffractometer HEiDi at the neutron source Forschungs-Neutronenquelle Heinz Maier-Leibnitz (FRM II). The sample was wrapped into aluminium foil and mounted to a closed cycle cryostat to reach a minimum temperature of 2.3 K (±0.1 K accuracy). Due to the large unit cell and the requested large Q range, a wavelength of 0.794 Å, in combination with a 15′ collimation of the incoming beam, was chosen. This setup allowed to measure all reflections in rocking scan mode (ω-scans) to suppress background modulation by powder circles from the sample environment (e.g. aluminium caps) and their contamination by neighbouring reflections. For the data collection itself, the hexagonal setting of the rhombohedral lattice was chosen. The lattice constants were refined from about 30 centred strong reflections (Table 2). At the beginning, a full data set without collimation (corresponding to about 60′ collimation) for the primitive cell was taken up to 2θ = 16°. From these 300 reflections, the profiles of a handful of them showed the occurrence of neighbouring reflections in their background. Switching to the 15′ collimation of the incoming beam was successfully tested to suppress this problem, and thus chosen for the following data sets in spite of the loss of neutron flux by a factor of four. The crystal was found to be affected by twinning by reticular merohedry (i.e. “obverse/reverse” twinning). All reflections followed the extinction rules expected for space group \({\it{R}} \overline{\text{3}} \) c (−h + k + l = 3n, obverse; h − k + l = 3n, reverse). Furthermore, only those reflections with l = 2n + 1 with l = 3n had significant intensities with I > 4σ(I). The goal of this experiment was to get as many significant non-symmetry-equivalent reflections up to large Q as possible. Therefore, in a further 15 days data collection, 7,115 reflections up to sin(θ)/λ = 0.91 Å−1 were measured (i.e. 1/6 shell, −h + k + l, along with at least one or two symmetry equivalent reflections), out of which 3,657 were unique for symmetry. The integrated intensities were corrected for Lorentz effect. Absorption correction was not applied because the weak absorption coefficient for neutrons (i.e. <0.01 cm−1) in pezzottaite. The discrepancy factor for the symmetry-related reflections was R int = 0.0523 (Table 2).
Structure refinement
The X-ray intensity data of pezzottaite were first processed with a series of programmes implemented in the WinGX package (Farrugia 1999), aimed to provide a valuable check of the distribution statistics of the normalized structure factors (E’s). The structure of pezzottaite was found being centrosymmetric at 83.5 % likelihood. A comparison of the equivalent reflections suggested the space group \({\it R} \overline{\text{3}} \) c as highly likely. The structure refinement (against F 2) was then performed with the SHELX-97 software (Sheldrick 1997, 2008), using as starting atomic coordinates those of the structural model of Yakubovich et al. (2009). The neutral X-ray scattering curves of Cs, Na, Li, Be, Al, Si and O were used according to the International Tables for Crystallography C (Wilson and Prince 1999). The secondary isotropic extinction effect was modelled according to the Larson’s formalism (Larson 1970). The first cycles of refinement were conducted with isotropic displacement parameters. According to the previous experimental findings, the Si1, Si2 and Si3 sites were modelled using the scattering curve of silicon, the Al1 and Al2 sites with the scattering curve of aluminium alone, the Li site with the curve of lithium and the Be site with the curve of beryllium, respectively. The channel content was modelled with the scattering curve of caesium at the Cs1 and Cs2 sites, and the curve of sodium at the Na1 and Na2 sites. The last cycles of refinement were conducted with anisotropic displacement parameters; only the Na1 and Na2 sites were modelled as isotropic, due to the significantly low site occupancy factor (s.o.f., Tables 3, 4 and 5). When convergence was achieved, the variance–covariance matrix showed no significant correlations amongst the refined parameters. At the end of the refinement, no peak larger than −0.5/+0.8 e − /Å3 was present in the final difference-Fourier map of the electron density (Table 2). We obtained a final agreement index R 1 = 0.0419 for 145 refined parameters and 2,155 unique reflections with Fo > 4σ(Fo) (Table 2). Atomic coordinates are listed in Table 3, and the displacement parameters are given in Tables 4, 5. A series of further test refinements were conducted with a different ordering scheme as follows:
-
1.
A mixed scattering curve of lithium + aluminium at the octahedral Al1, Al2 and Al3 sites.
-
2.
A mixed scattering curve of lithium + beryllium at the tetrahedral Li and Be sites.
However, no improvement in the figure of merit of the refinement was observed with lithium at the octahedral sites (i.e. the refinement converged with aluminium alone at the octahedral sites), and neither with a disordered lithium/beryllium distribution at the two tetrahedral sites.
The neutron intensity data were processed with the same protocol used for the X-ray refinement. The preliminary data treatment suggested that the structure is centrosymmetric at 90.5 % likelihood. The structural refinement (against F) was then performed with Jana2006 software (Petricek et al. 2006), starting from the X-ray structural model previously refined. The neutron scattering lengths of Cs, Na, Li, Be, Al, Si and O according to Sears (1986) were used. The secondary isotropic extinction effect was corrected according to the formalism of Becker and Coppens (1974), as implemented in Jana2006. As the crystal was found to be affected by twinning (by reticular merohedry), the refined volume fractions of the two individuals (obverse/reverse) were 46.5(5) and 53.5(5) %, respectively. The neutron refinement was conducted using isotropic displacement parameters for all the atomic sites. Only the channel Cs1 and Cs2 sites were refined anisotropically. The use of anisotropic displacement parameters for the other atomic sites did not improve the figures of merit of the refinement. Likely, the low T at which the neutron diffraction data were collected (i.e. 2.3 K) leads to an appropriate description of the atomic thermal motion with an isotropic vibrational model. However, with a fully ordered distribution of lithium and beryllium at the tetrahedral Li and Be sites, respectively, the refinement did not achieve the convergence, with significant minima/maxima at the site coordinates of the Li and Be sites. Further refinement cycles were then performed assuming a disordered distribution of lithium and beryllium at both Li and Be sites. With such a configuration, convergence was rapidly achieved. No evidence of lithium/aluminium disordering at the octahedral sites was observed. A careful inspection of the difference-Fourier map of the nuclear density did not show evidence of oxygen or hydrogen sites, ascribable to H2O molecules, as channel content. At the end of the refinement, no peak larger than ~−2.0/+2.5 fm/Å3 was present in the difference-Fourier map of the nuclear density (Table 2), and the variance–covariance matrix showed no significant correlation amongst the refined parameters. The final agreement index (R 1) was 0.0595 for 70 refined parameters and 1,818 unique reflections with I > 3σ(I) (Table 2). The refined atomic positions and displacement parameters are listed in Table 6.
Bond lengths obtained by the X-ray and neutron structure refinements are given in Table 7.
IR spectroscopy
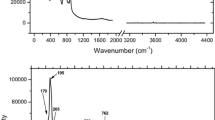
The infrared spectrum of the pezzottaite here analyzed shows two weak absorption bands at 3,591 and 3,545 cm−1, which can be assigned to the fundamental H2O stretching vibrations (Fig. 2), as previously observed by Hawthorne et al. (2004) and Liu and Peng (2005). On the basis of the H2O frequencies in beryl, the first absorption band can be assigned to ν1 (symmetric stretching) mode of H2O molecules, whose H···H vector is normal to [0001] (“type II”) (Wood and Nassau 1967, 1968; Aines and Rossman 1984; Aurisicchio et al. 1994; Charoy et al. 1996; Fukuda and Shinoda 2011). On the contrary, the 3,545-cm−1 band was never reported for beryl. Its assignment is still dubious, though likely ascribable to a further stretching mode of H2O. Four distinct and pronounced absorption bands occur between 1,120 and 920 cm−1 (i.e. 1,117, 1,040, 952 and 924 cm−1, Fig. 2), which can be assigned to Si–O stretching modes (Farmer 1974; Hofmeister et al. 1987; Aurisicchio et al. 1994). The 1,117-cm−1 absorption band is shifted towards lower frequencies with respect to the co-respective one in beryl (i.e. ~1,200 cm−1) upon the replacement of beryllium by lithium, according to Yakubovich et al. (2009). The absorption bands observed between 840 and 700 cm−1 (i.e. 831, 751 and 700 cm−1, Fig. 2) can be tentatively assigned to the Be–O and Li–O stretching modes, coupled with O–Si–O bending modes (Farmer 1974; Hofmeister et al. 1987, Aurisicchio et al. 1994; Prencipe et al. 2006). In their comparative study on the crystal chemistry and IR spectra of natural beryls, Aurisicchio et al. (1994) highlighted how the absorption band at 700 cm−1 is strongly influenced by the presence of lithium, which replaces beryllium in tetrahedral coordination (i.e. the higher the Li content, the higher the absorption), coupled with a shift towards higher frequencies of the band at ~800 cm−1. Two-weak absorption bands are observed at 638 and 624 cm−1, and can be assigned to O–Be–O and O–Al–O bending modes (Prencipe et al. 2006).

The FT infrared spectrum of pezzottaite in the regions 600–1,200 cm−1 (above) and 3,400–3,800 cm−1 (below), in transmission mode by KBr pellet. Note the different vertical scale
Discussion and conclusions
In this study, the crystal structure and crystal chemistry of pezzottaite were investigated, for the first time, by single-crystal X-ray and neutron diffraction, IR spectroscopy, EMPA-WDS and TG analysis.
The EMPA-WDS analysis shows a chemical homogeneity of the sample of pezzottaite used in this study (Table 1), without evidence of zoning. The average chemical formula of the pezzottaite sample (assuming the Li and Be fraction deduced on the basis of the single-crystal neutron structure refinement) is the following:
We expect that (a) rubidium and potassium can share the Cs1 and Cs2 sites along with caesium, (b) calcium can share the Na1 and Na2 channel sites with sodium and (c) magnesium, manganese and iron share the Al1 and Al2 sites along with aluminium. In all the chemical analyses of pezzottaite previously reported, the Cs content appears being systematically lower than 1 a.p.f.u. (Laurs et al. 2003; Simmons et al. 2003; Yakubovich et al. 2009). Also in this case, we observe partial site occupancy of the channel Cs1 and Cs2 sites.
The (unpolarized) IR spectrum over the region 3,800–600 cm−1 collected in this study is similar to those previously reported (Hawthorne et al. 2004; Liu and Peng 2005; Yakubovich et al. 2009), and shows two weak absorption bands ascribable to the fundamental H2O stretching vibrations (i.e. 3,591 and 3,545 cm−1, Fig. 2). It appears, therefore, that the H2O content in our pezzottaite is modest, as reflected by the TG results (0.4 wt%, 0.27 m.p.f.u., Table 1). If we compare the IR spectrum of pezzottaite with those of Cs/Li-free beryl previously reported, we find that several of the bands ascribable to the tetrahedral Si–O and Be–O stretching and O–Si–O bending modes are significantly affected by the lithium/beryllium replacement in tetrahedral coordination (Farmer 1974; Hofmeister et al. 1987; Aurisicchio et al. 1994; Prencipe et al. 2006).
The single-crystal X-ray and neutron structure refinements show a general structure model of pezzottaite in agreement to that of Yakubovich et al. (2009). In our pezzottaite, the X-ray and neutron structure refinements show the presence of four independent channel sites (i.e. Na1, Na2, Cs1 and Cs2, Tables 3 and 6) mainly populated by monovalent cations. No significant evidence of H2O sites was observed. This reflects the low amount of H2O deduced on the basis of the TG analysis. Without H2O sites, the bonding configuration of the extra-framework population is summarized in Table 7: Cs1 and Cs2 sites are coordinated by 12 framework oxygen atoms (6 above and 6 below the cation), whereas the coordination shell of both Na1 and Na2 is represented by 6 oxygen sites almost co-planar with the cations (Fig. 1). The bonding configuration of the channel cations leads to a “pillar” running along [0001], as shown in Fig. 1.
The significantly different neutron scattering lengths of lithium (b c(Li) = −1.90 fm), beryllium (b c(Be) = 7.79 fm), silicon (b c(Si) = 4.1507 fm) and aluminium (b c(Al) = 3.449 fm) provide a clear picture about the site population at the tetrahedral (i.e. Li, Be, Si1, Si1, Si2 and Si3) and octahedral (i.e. Al1, Al2 and Al3) sites. The neutron refinement showed, unambiguously, (a) a non-significant presence of aluminium, beryllium or lithium at the Si1, Si2 and Si3 sites, (b) the absence (at a significant level) of lithium at the octahedral Al1, Al2 and Al3 sites, (c) a partial lithium/beryllium disordering between tetrahedral Be and Li sites (Table 6). In particular, the neutron structure refinement converges with a population of the Be site represented by (96.5(6)% Be + 3.5(6)% Li), whereas the population of the Li site is represented by (14.8(6)%Be + 85.2(6)%Li) (Table 6). This finding is not surprising if we consider that Cs/Li-rich beryls (e.g. “vorobyevite”, Yakubovich et al. 2009) have a disordered Be/Li distribution.
The neutron structure refinement provides a total amount of beryllium and lithium of 2.078(18) and 0.922(18) a.p.f.u., respectively (Table 6). This small, but significant, deficit of lithium was actually reported in all the chemical analysis of pezzottaite previously published (Laurs et al. 2003; Simmons et al. 2003; Hawthorne et al. 2004; Yakubovich et al. 2009). On this basis, a Li/Be disorder between Be and Li sites in pezzottaite structure is expected. It appears, therefore, that the change in symmetry between beryl (i.e. P6/mcc) and pezzottaite (i.e. \({\it{R}} \overline{\text{3}} \) c) cannot be ascribed only to the Li/Be ordering, but more likely to the nature and bonding configuration of the channel content. On the basis of our experimental finding, we suggest that the general formula of pezzottaite should be written as: (Cs,Na)1−x (Be2+x Li1−x )Al2Si6O18 (with x < 0.2).
The discrepancy between the X-ray and neutron structure refinement about the occupancy factor of the Cs1 and Cs2 sites (Tables 3 and 6) is likely due to the multi-element population of the channel sites. Using the scattering curve of caesium alone, the X-ray refinement converges with s.o.f.(Cs1) = 0.694(1) and s.o.f.(Cs2) = 0.764(1) (i.e. total of 0.72 a.p.f.u.), whereas the use of caesium neutron scattering length leads the neutron refinement to converge with s.o.f.(Cs1) = 0.95(2) and s.o.f.(Cs2) = 0.98(1) (total of 0.96 a.p.f.u.). Such a discrepancy could be explained by the use of scattering curve/length of caesium alone to model the Cs1 and Cs2 sites: the presence of lighter elements (i.e. Rb, K or Ca) would make the “aggregate” X-ray s.o.f. higher than those we obtained. However, the discrepancy between the chemical analysis, in which (Cs + Rb + K) = 0.61 a.p.f.u. (Table 1), and the results of the refinements is significant. But if we consider the possibility that H2O molecules replace Cs (along with Rb and K) at Cs1 at Cs2 sites or Na (and Ca) at Na1 and Na2 sites, we obtain a total number of electrons for the channel populations, deduced on the basis of the chemical analysis, of 36.2 e − p.f.u. vs. 41.1 e − p.f.u. by X-ray structure refinement. In this case, the difference in electrons is reduced to ~12 %. In other words, we cannot exclude a disordered distribution of cations and H2O molecules between the Cs1,Cs2, Na1 and Na2 sites. A further evidence of the presence of H2O at the aforementioned cation sites is provided by the neutron structure refinement: the Cs1–Na1, Cs–Na2 and Cs2–Na2 distances are all shorter than 2.5 Å. Thus, Cs1 and Na1, Cs1 and Na2 and Cs2 and Na2 are mutually exclusive, if we consider only a cationic population. However, the refined s.o.f. using the scattering lengths of caesium at Cs1 and Cs2 and that of sodium at Na1 and Na2, respectively, led to unrealistic: s.o.f.(Cs1) + s.o.f.(Na1) = 1.14(6), s.o.f.(Cs1) + s.o.f.(Na2) = 1.07(6) and s.o.f.(Cs2) + s.o.f.(Na2) = 1.10(7) (Table 6). If H2O partially substitutes Cs, the corrected s.o.f. would lead to a realistic bonding scenario with, for example, NaO6OH2 polyhedra. The similar neutron scattering length of Cs and O (i.e. b c(Cs) = 5.42 fm, b c(O) = 5.805 fm) did not allow an unambiguous refinement of the O and Cs fraction at the Cs1 and Cs2 sites. However, a “virtual” excess of caesium (or sodium, i.e. b c(Na) = 3.63 fm) can be easily explained by the presence of oxygen sharing the same site.
The anisotropic X-ray structural refinement shows that the displacement ellipsoids are only slightly pronounced for all the atomic sites (Table 5). Only for the O3, O5 and O9 sites, a slightly pronounced anisotropy is observed. This is not surprising if we consider that O3 and O5 are both coordinated by the Li site, and so the displacement around the centre of gravity can be due to the lithium/beryllium disorder. The O9 site is the bridging oxygen between the Si2 and Si3 tetrahedra, and it belongs to the coordination shell of Cs1 and Cs2 as well. Such a complex bonding configuration can explain its slightly pronounced displacement anisotropy.
Despite the beryllium/lithium disorder at the Be and Li sites, the coordination tetrahedra appear to be only slightly distorted (Table 7), as shown by the Δ(T–O)max (i.e. the difference between the longest and the shortest bond distances) in both X-ray and neutron refinement: Δ(Li–O)max ~ 0.005 Å and Δ(Be–O)max ~ 0.025 Å for X-ray data, Δ(Li–O)max ~ 0.017 Å and Δ(Be–O)max ~ 0.019 Å for neutron data.
The tetrahedral and octahedral bond distances were corrected for “rigid body motions” following the protocol of Downs et al. (1992) and Downs (2000) (Table 7). If we consider the X-ray structure refinement, based on data collected at 298 K, the difference between the average (uncorrected) polyhedral bond distances and the co-respective corrected for “rigid body motion” is all less than 0.008 Å, reflecting a modest magnitude of the site displacement parameters as previously discussed. The only exception is represented by the Li tetrahedron, for which the difference is 0.035 Å, somehow reflecting the pronounced displacement regime of the coordinated oxygen sites (i.e. O3 and O5). In general, the difference between the uncorrected and corrected bond distances for “rigid body motion” is reduced in the neutron refinement, based on data collected at 2.3 K.
References
Agilent Technologies (2012) Xcalibur CCD system, CrysAlis Software system
Aines RD, Rossman GR (1984) The high-temperature behaviour of water and carbon dioxide in cordierite and beryl. Am Mineral 69:319–327
Armbruster T, Libowitzky E, Diamond L, Auernhammer M, Bauerhansl P, Hoffmann C, Irran E, Kurka A, Rosenstingl H (1995) Crystal chemistry and optics of bazzite from Furkabasistunnel (Switzerland). Min Petrol 52:113–126
Armstrong JT (1988) Quantitative analysis of silicate and oxide materials: comparison of Monte Carlo, ZAF, and phi(rho Z) procedures. In: Newbury DE (ed) Microbeam analysis. San Francisco Press, USA, pp 239–246
Aurisicchio C, Fioravanti G, Grubessi O, Zanazzi PF (1988) Reappraisal of the crystal chemistry of beryl. Am Mineral 73:826–837
Aurisicchio C, Grubessi O, Zecchini P (1994) Infrared spectroscopy and crystal chemistry of the beryl group. Can Min 32:55–68
Becker PJ, Coppens P (1974) Extinction within the limit of validity of the Darwin transfer equations. I. General formalism for primary and secondary extinction and their applications to spherical crystals. Acta Cryst A30:129–147
Černý P (2002) Mineralogy of beryllium in granitic pegmatites. In Grew ES (ed) Beryllium: mineralogy, petrology, and geochemistry. Reviews in Mineralogy and Geochemistry, Mineralogical Society of America, Washington DC, vol 50, pp 405–444
Černý P, Anderson AJ, Tomascak PB, Chapman R (2003) Geochemical and morphological features of beryl from the Bikita granitic pegmatite, Zimbabwe. Can Min 41:1003–1011
Charoy B, De Donato P, Barres O, Pinto-Coelho C (1996) Channel occupancy in alkali-poor beryl from Serra Branca (Goias, Brasil): spectroscopic characterization. Am Mineral 81:395–403
Della Ventura G, Rossi P, Parodi GC, Mottana A, Raudsepp M, Prencipe M (2000) Stoppaniite, (Fe,Al,Mg)4(Be6Si12O36) • (H2O)2(Na,□) a new mineral of the beryl group from Latium (Italy). Eur J Mineral 12:121–127
Downs RT (2000) Analysis of harmonic displacement factors. In: Hazen RM, Downs RT (eds) High-temperature and high-pressure crystal chemistry, vol 41. Reviews in Mineralogy and Geochemistry, Mineralogical Society of America and Geochemical Society, Washington, USA, pp 61–117
Downs RT, Gibbs GV, Bartelmehs KL, Boisen Jr MB (1992) Variations of bond lengths and volumes of silicate tetrahedra with temperature. Am Mineral 77:751–757
Farmer VC (1974) The infrared spectra of minerals. Mineralogical Society, London
Farrugia LJ (1999) WinGX suite for small-molecule single-crystal crystallography. J Appl Crystallogr 32:837–838
Ferraris G, Prencipe M, Rossi P (1998) Stoppaniite, a new member of the beryl group: crystal structure and crystal-chemical implication. Eur J Mineral 10:491–496
Fukuda J, Shinoda K (2011) Water molecules in beryl and cordierite: high-temperature vibrational behavior, dehydration, and coordination to cations. Phys Chem Minerals 38:469–481
Gatta GD, Nestola F, Bromiley GD, Mattauch S (2006) The real topological configuration of the extra-framework content in alkali-poor beryl: a multi-methodological study. Am Mineral 91:29–34
Gatta GD, Rotiroti N, Fisch M, Kadiyski M, Armbruster T (2008) Stability at high-pressure, elastic behaviour and pressure-induced structural evolution of CsAlSi5O12, a potential nuclear waste disposal phase. Phys Chem Minerals 35:521–533
Gatta GD, Rinaldi R, McIntyre GJ, Nénert G, Bellatreccia F, Guastoni A, Della Ventura G (2009a) On the crystal structure and crystal chemistry of pollucite, (Cs, Na)16Al16Si32O96·nH2O: a natural microporous material of interest in nuclear technology. Am Mineral 94:1560–1568
Gatta GD, Rotiroti N, Boffa Ballaran T, Sanchez Valle C, Pavese A (2009b) Elastic behaviour and phase-stability of pollucite, a potential host for nuclear waste. Am Mineral 94:1137–1143
Gatta GD, Vignola P, McIntyre GJ, Diella V (2010) On the crystal chemistry of londonite [(Cs,K,Rb)Al4Be5B11O28]: a single-crystal neutron diffraction study at 300 and 20 K. Am Mineral 95:1467–1472
Hawthorne FC, Cooper M, Simmons WB Jr, Laurs BM, Armbruster Th, Rossman GR, Peretti A, Günter D, Grobéty B, Falster AU (2004) Pezzottaite Cs(Be2Li)Al2Si6O18: a spectacular new beryl-group mineral from the sakavalana pegmatite, Fianarantsoa Province, Madagascar. Mineral Rec 35:369–378
Hofmeister AM, Hoering TC, Virgo D (1987) Vibrational spectroscopy of beryllium aluminosilicates: heat capacity calculations from band assignments. Phys Chem Mineral 14:205–224
Larson AC (1970) Secondary-extinction corrections. In: Ahmed FR, Hall SR, Huber CP (eds) Crystallographic computing. Munksgaard Publisher, Copenhagen, Denmark, pp 291–294
Laurs BM, Simmons WB, Rossman GR, Quinn EP, McClure SF, Pereiti A, Armbruster Th, Hawthorne FC, Falster AU, Günther D, Cooper MA, Grobéty B (2003) Pezzottaite from Ambatovita, Madagascar: a new gem mineral. Gems Gemol 39:284–301
Liu SI, Peng MS (2005) A new gem mineral. The vibrational spectroscopic characterization of pezzottaite. Acta Mineral Sin 25:60–64
Petricek V, Dusek M, Palatinus L (2006) Jana2006. The crystallographic computing system. Institute of Physics, Praha, Czech Republic
Prencipe M, Noel Y, Civalleri B, Roetti C, Dovesi R (2006) Quantum-mechanical calculation of the vibrational spectrum of beryl (Al4Be6Si12O36) at the Γ point. Phys Chem Minerals 33:519–532
Sears VF (1986) Neutron scattering lengths and cross-sections. In: Sköld K, Price DL (eds) Neutron scattering, methods of experimental physics, vol 23A. Academic Press, New York, pp 521–550
Sheldrick GM (1997) SHELX-97. Programs for crystal structure determination and refinement. University of Göttingen, Germany
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A64:112–122
Simmons WB, Falster AU, McClure SF, Quinn EP, Rossman GR, Hawthorne FC (2003) Gem news international: a new saturated purplish pink Cs-“beryl” from Madagascar: preliminary analyses. Gems Gemol 39:50–54
Wilson AJC, Prince E (eds) (1999) International tables for X-ray crystallography, Volume C: mathematical, physical and chemical tables, 2nd edn. Kluwer Academic, Dordrecht, NL
Wood DL, Nassau K (1967) Infrared spectra of foreign molecules in beryl. J Chem Phys 42:2220–2228
Wood DL, Nassau K (1968) The characterization of beryl and emerald by visible and infrared absorption spectroscopy. Am Mineral 53:777–800
Yakubovich OV, Pekov IV, Steele IM, Massa W, Chukanov NV (2009) Alkali metals in beryl and their role in the formation of derivative structural motifs: comparative crystal chemistry of vorobyevite and pezzottaite. Crystallogr Rep 54:399–412
Acknowledgments
The authors thank the Forschungs-Neutronenquelle Heinz Maier-Leibnitz (FRM II), München, Germany, for the allocation of neutron beam time. Dr. F. Pezzotta is acknowledged for providing the sample of pezzottaite here investigated. S. Mills, an anonymous reviewer and the Editor, M. Rieder, are thanked.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gatta, G.D., Adamo, I., Meven, M. et al. A single-crystal neutron and X-ray diffraction study of pezzottaite, Cs(Be2Li)Al2Si6O18 . Phys Chem Minerals 39, 829–840 (2012). https://doi.org/10.1007/s00269-012-0539-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-012-0539-3