
Abstract
We report on high-pressure and high-temperature experiments involving carbonates and silicates at 30–80 GPa and 1,600–3,200 K, corresponding to depths within the Earth of approximately 800–2,200 km. The experiments are intended to represent the decomposition process of carbonates contained within oceanic plates subducted into the lower mantle. In basaltic composition, CaCO3 (calcite and aragonite), the major carbonate phase in marine sediments, is altered into MgCO3 (magnesite) via reactions with Mg-bearing silicates under conditions that are 200–300°C colder than the mantle geotherm. With increasing temperature and pressure, the magnesite decomposes into an assemblage of CO2 + perovskite via reactions with SiO2. Magnesite is not the only host phase for subducted carbon—solid CO2 also carries carbon in the lower mantle. Furthermore, CO2 itself breaks down to diamond and oxygen under geotherm conditions over 70 GPa, which might imply a possible mechanism for diamond formation in the lower mantle.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recent studies indicate that subducted oceanic plates stagnate around the transition zone (660 km discontinuity) because of buoyancy effects; parts of the stagnant slabs sink into the lower mantle after growing to the critical size of a megalith (Ringwood and Irifune 1988) or following transformation to a perovskite lithology (Hirose et al. 1999). The amount of carbonate within marine sediment that is subducted at convergent margins is estimated to be as much as 1.8 × 1012 mol/year (Sano and Williams 1996); this represents an important problem in the global recycling of carbon in terms of the depth to which the carbonates are transported into the Earth’s interior. Previous experimental studies indicate the following scenario. In basaltic compositions, CaCO3 (calcite and aragonite, the most abundant carbonates within marine sediments) is transformed to CaMg[CO3]2 (dolomite) and MgCO3 (magnesite) at pressures up to ∼3 GPa (Yaxley and Brey 2004). CaMg[CO3]2 itself breaks down to a CaCO3 + MgCO3 assemblage at pressures between 5 and 8 GPa (Morlidge et al. 2006; Sato and Katsura 2001; Luth 2001). In the absence of any silicates, MgCO3 alone is the stable solid phase under all mantle conditions irrespective of phase transitions (Biellmann et al. 1993; Isshiki et al. 2003). Although uncertainty remains as to whether carbonatitic melts are produced in basalt + calcite composition along subduction geotherms in the upper mantle (Hammouda 2003; Yaxley and Brey 2004; Dasgupta et al. 2005), it is not necessarily the case that all hosts of subducted carbon are removed from the subducted slabs before reaching the lower mantle. Accordingly, magnesite appears to be the major host of carbon in the deep mantle. Recent work (Takafuji et al. 2006), however, has demonstrated that magnesite can decompose into solid CO2 in the presence of SiO2, which is a major phase of basaltic composition within the lower mantle. Thus, solid CO2 also might be a host of subducted carbon, although the decomposition reaction has not been confirmed in a system that resembles the natural system. In this study, we demonstrate the further fate of subducted carbonates in a system analogous to carbonated oceanic crust.
Experiments
We used a mixture of 80 wt% MORB glass and 20 wt% CaCO3 calcite (Cc) as a starting material, which represents a simplified model of carbonated oceanic crust. Samples of MORB glass were prepared using a drop–quench technique after melting the oxide mixture at 1,523 K in a controlled oxygen fugacity using an electric furnace with a flow system of mixed H2/CO2 gases (fO2 = 1 × 10−9 atm) so that all iron of the starting material is ferrous (Fe2+). The composition of the glass was measured using electron probe micro-analysis (EPMA). The oxide totals (Na2O: 2.52, MgO: 7.91, Al2O3: 15.76, SiO2: 51.58, CaO: 11.33, TiO2: 1.13, MnO: 0.20, FeO: 9.56, Total: 100.2 in wt%) were very close to 100 wt%, and the composition was found to be similar to average MORB composition (Perterrmann and Hirschmann 2003). It should be noted that measured carbonate/silicate ratios of the oceanic crust vary widely among different sites. We selected the above ratio not as a realistic model but to facilitate identification of the product phases derived from the carbonate. We assume that the mixture of basalt and carbonate is able to sink into the lower mantle while retaining the original bulk composition.
High-pressure and high-temperature experiments were carried out in a symmetrical diamond anvil cell (DAC, Syntek, Japan) with a YLF laser-heating system. The mixed samples were sandwiched between NaCl pellets, loaded into a hole (100 μm diameter) within a rhenium gasket, and pressed by 200–300 μm culet diamond anvils. NaCl works as both a pressure-transmitting medium and a thermal insulator. The sample was heated from both sides using a YLF laser (TEM01* mode) and reacted in a given area for 1–2 h. The temperature was measured from one side according to the blackbody radiation law on the basis of thermal radiation. The laser spot size was about 20 μm and temperature uncertainty was about ±200 K. The Raman shift of diamond was used as a pressure scale (Akahama and Kawamura 2004). Uncertainty in the measured pressures was about 5 GPa; this does not include a correction for thermal pressures. For phase identification of the reacted samples, we used both synchrotron X-ray diffraction (SR-XRD) and analytical transmission electron microscopy (ATEM). SR-XRD measurements were carried out on beamlines BL13A at PF-KEK in Tsukuba, Japan, and BL10XU at SPring-8 in Hyogo, Japan. The incident X-ray beam was monochromatized to wavelengths of 0.6177 (PF-KEK) and 0.4153 Å (SPring-8) and collimated to 20 μm in diameter; the diffracted beam was detected using a flat imaging plate (IP, Rigaku R-AXIS IV). Two-dimensional X-ray diffraction patterns were analysed using software (IPAnalyzer and PDIndexer) developed by the authors (http://mineralx.sci.hokudai.ac.jp/~seto/). Chemical and microstructural analyses of the recovered samples were undertaken using an ATEM (JEOL JEM-2010) equipped with a LaB6 cathode, operated at 200 kV. ATEM specimens were thinned by Ar ion milling after trimming off the insulation layers. Chemical compositions were measured using an energy-dispersive analytical system (EDS, Thermo Electron. Noran System SIX) attached to an ATEM.
Results
Table 1 lists the P–T conditions and run products used in this study. A total 35 runs were performed at 30–80 GPa and 1,600–3,200 K, corresponding to depths within the Earth of approximately 800–2,200 km. All run products were investigated by SR-XRD and/or ATEM.
At pressures less than 60 GPa and temperatures less than 2,000 K (run nos. 1–6), the reaction products consisted of MgSiO3-perovskite (Mg-Pv), CaSiO3-perovskite (Ca-Pv), Stishovite (St), CaFe2O4-type aluminous phase (CF), magnesite (Mgs), and aragonite (Arg), as shown in Figs. 1a, b. ATEM observations were used to identify the carbonate phases (Mgs and Arg). The CaCO3 component in the starting material was provided by crystalline calcite (diameter ∼1 μm), ensuring that realistically small particles of CaCO3 were distributed throughout the sample chamber. Consequently, the CaCO3-rich area gave rise to Ca-Pv + St + CF + Mgs + Arg (Fig. 1b) and the CaCO3-poor area yielded Ca-Pv + Mg-Pv + St + CF + Mgs. TEM observations also revealed that Mg-Pv grains were not in contact with Arg grains within the laser-heated area. The product phases of calcite-free MORB glass under the same conditions were Mg-Pv + Ca-Pv + St + CF, which are consistent with the findings of previous studies (Hirose et al. 2005). On the basis of these results, the following reaction is considered to be dominant for the tested calcite-bearing system under pressure conditions below 60 GPa and temperatures below 2,000 K:
Although Reaction 1 has been reported previously in the MgSiO3–CaCO3 system (Biellmann et al. 1993), we confirmed it in a system that resembles the natural system. At pressures of 60–80 GPa and temperatures of less than 2,300 K (run nos. 7–8), the same assemblages were observed except for the phase transition from stishovite to the CaCl2-structured SiO2 phase (CS).

TEM images of the reaction products of MORB + CaCO3 runs recovered from a, b 44 GPa and 1,900 K and c 42 GPa and 3,000 K; d is the electron diffraction pattern for the diamond shown in c
At higher temperatures than 2,000 K, discontinuous changes in temperature were observed during the laser heating: after a slight increase in laser power around 2,000 K at 60 GPa or less, the temperature increased rapidly to 2,850 K or higher (run nos. 9–13). Also, on decreasing laser power after discontinuous changes, temperature discontinuously decreased. On the other hand we were able to smoothly control the temperature at pressures above 60 GPa (run nos. 14–15). At pressures of less than 60 GPa and temperatures above 2,850 K (run nos. 9–13), the run products changed to Mg-Pv, Ca-Pv, St, CF, and diamond (Figs. 1c, d); no carbonates were observed in the reacted zones. Diamond was also observed at pressures of 60–80 GPa and temperatures above 2,400 K (run nos. 14–15). The diamond grains are relatively large in size (∼1 μm) and smaller in number than the other constituent phases. The electron diffraction pattern (Fig. 1d) is fully consistent with that of cubic diamond \( (Fd\ifmmode\expandafter\bar\else\expandafter\=\fi{3}m). \) Our current interpretation is that the following reactions occurred in succession:
At 30–80 GPa and around 2,000 K, the non-molecular solid phase (CO2-V or CO2-VI) is the possible form of CO2 (Iota and Yoo 2001; Tschauner et al. 2001; Yoo et al. 1999). Oxygen probably exists as the ε-phase (Akahama et al. 1995) under the conditions of the lower mantle. The exact temperature at which Reactions 2 and 3 began in the studied system could not be determined because of the discontinuous changes in temperature observed during the experiments.
Because Mg-Pv is a primary high-pressure phase in the carbonate-free MORB composition, it is difficult to directly verify Reactions 2 and 3 in the above system. Accordingly, we conducted additional experiments using a simplified system, SiO2 + MgCO3 (1:1 mol ratio), as shown in Table 1. SiO2 is a reagent α-quartz. MgCO3 is natural magnesite with a composition of Mg0.99Fe0.01CO3, from Bahia, Brazil. The sample was mixed with platinum powder as a laser-radiation absorber. The mixed ratios of platinum were 5 wt% for run nos. 20–26 and 30–35, and 50 wt% for run nos. 27–29. In the system with a small amount of platinum, a dramatic increase in temperature was observed at pressures of less than 60 GPa (run nos. 31–32); the critical temperatures were similar to those observed in the MORB + CaCO3 experiments. In the system with a large amount of platinum (run nos. 27–29), however, the dramatic temperature increase was not observed, whereas pressures were less than 60 GPa. X-ray diffraction patterns (Fig. 2) revealed that the reaction products prior to the temperature changes consisted of only Mgs + St, while Mg-Pv was found in the reaction products after the changes. Therefore, it is considered that the CO2 release reaction (Reaction 2) occurs above the critical temperatures. TEM observations also reveal diamonds in the reaction products following the changes in temperature. As with the earlier experiments, we were able to smoothly control temperature at pressures above 60 GPa, irrespective of the amount of platinum. The reaction products consisted of only Mgs + SiO2 at pressures between 29 and 77 GPa and temperatures less than 2,000 K (run nos. 20–26), while diamond and Mg-Pv were observed at 31 GPa and 3,300 K (run no. 31), 44 GPa and 3,300 K (run no. 32), and 73 GPa and 2,500 K (run no. 33). At pressures between 45 and 69 GPa and temperatures between 2,300 and 2,500 K (run nos. 27–30), we failed to observe diamond but detected Mg-Pv in the reaction area. This result suggests that Reaction 2 takes place under these conditions but Reaction 3 does not.

X-ray diffraction profiles for the samples of the SiO2 + MgCO3 runs recovered from a 42 GPa and 3,300 K and b 43 GPa and 1,900 K. Platinum was used as a laser-radiation absorber
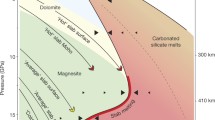
To further verify the preferred occurrence of Reactions 2 and 3, we examined the thermodynamic properties of the probable phase assemblages (Yoo et al. 1999; Akahama et al. 1995; Andrault et al. 2000; Occelli et al. 2003; Fiquet et al. 2002; Shim and Duffy 2000), as shown in Fig. 3. We compared the molar volumes at 300 K because the volume data of CO2 and O2 at high temperatures are not available. The assemblage of Mg-Pv + CO2-V has smaller molar volumes than Mgs + St has over the entire range of the lower mantle although the volume property of CO2-VI, a higher pressure phase than CO2-V, remains unresolved. Furthermore, the volume of the mixture of diamond + ε-O2 is smaller than that of any other forms of CO2 above about 22 GPa. Accordingly, the successive Reactions 2 and 3 appear to be thermodynamically favourable under high-pressure conditions.

Molar volumes for the assemblages of magnesite (Fiquet et al. 2002) + stishovite (Andrault et al. 2000), Mg-perovskite (Shim and Duffy 2000) + CO2 (V) (Yoo et al. 1999), and Mg-perovskite + diamond (Occelli et al. 2003) + ε-O2 (Akahama et al. 1995) under 300 K isothermal conditions. The assemblage of Mg-perovskite + diamond + ε-O2 has smaller molar volumes than the other two assemblages have at pressures above ∼23 GPa
Discussion
Figure 4 summarizes the experimental conditions and results obtained in this study. Lines for reactions 2 and 3 proposed in this study are also shown. According to the volume calculations, Reactions 2 and 3 would proceed with increasing pressures at fixed temperatures. Takafuji et al. (2006) reported that Reaction 2 occurs in a SiO2 + MgCO3 system at temperatures much lower than those proposed in the current study. Moreover, they did not refer to discontinuous changes in temperature. The discontinuous changes were not observed in the carbonate-free MORB system or silicate-free magnesite system under the same conditions as shown in Table 1. In the current study, discontinuous temperature changes were observed in a system without platinum (run nos. 9–13) and with a small amount (5 wt%) of platinum (run nos. 31–32), whereas the phenomenon was not observed in a system with a large amount (50 wt%) of platinum (run nos. 27–29). The mixed ratio of platinum in Takafuji et al. (2006) was also relatively large (10 wt%). Therefore, the different amounts of the mixed platinum may cause the different results between Takafuji et al. (2006) and the current study. Moreover, we should say that vapour (especially H2O) contamination may affect the reaction temperatures and the phenomenon of the discontinuous temperature changes. In the current study, carbonates and NaCl, relatively hygroscopic material, were used. It is well known that even a small amount of water brings down the melting temperature in most silicate systems. Hirose et al. (1999) has reported that the phenomenon of the discontinuous changes in temperature observed during LH-DAC experiments is the result of the latent heat of melting. Therefore, the discontinuous changes observed in the current study might be due to partial melting of the sample, although we were unable to find any evidence of melting such as quenching texture, even under TEM observations. If partial melting had occurred, only a small amount of melting is inferred to have occurred. Further investigations of the phenomenon of the discontinuous changes in temperature are therefore required.

Experimental conditions and results for the current study and Takafuji et al. (2006). Solid symbols indicate that diamonds are confirmed in the reacted zone. The typical mantle geotherm (broad gray line, Stacey 1992) and the decomposition boundary of CO2 by Tschauner et al. (2001) (gray dashed line) are also shown. Solid reaction lines are those proposed in the current study
Subducted slabs should be colder than the surrounding mantle. If the subducted slabs within the lower mantle are 200–300°C colder than the surrounding mantle, magnesite in the subducted slabs is stable and is likely to be delivered deep into the lower mantle, as shown in Fig. 4. In contrast, if the subduction geotherm is similar to the mantle geotherm (i.e., the rate of subduction is lower), magnesite is unstable and the decarbonation reaction may proceed in the presence of SiO2 according to Reaction 2 as shown in Fig. 4. Therefore, magnesite is not the only host phase for subducted carbon in the lower mantle, as noted in Takafuji et al. (2006); in contrast with the findings of previous studies (Biellmann et al. 1993; Isshiki et al. 2003), solid CO2 also carries subducted carbon. Furthermore, we obtained the first experimental evidence that carbonate within subducted slabs decomposes into diamond. Following the decarbonation reaction described in Reaction 2, carbon dioxide itself breaks down to diamond and oxygen via Reaction 3. It is doubtful that after Reaction 3 free oxygen remains stable under actual mantle conditions. The presence of solid O2 is not warranted because of the presence of Mg-Pv in the MORB system. Mg-Pv can dissolve a considerable amount of Fe3+ (Nishio-Hamane et al. 2005). The oxygen might work as the oxidant and would no longer exist in this case. Tschauner et al. (2001) have reported that solid CO2 decomposes to oxygen and diamond along a boundary having negative P–T slope above 30 GPa and moderate temperatures. The current data suggest that the Reaction 3 occurs at temperatures higher than that inferred from Tschauner et al. (2001) as shown in Fig. 4, although the exact critical temperature of decomposition of carbon dioxide at lower pressures (<70 GPa) is uncertain because of the rapid increase in temperature during laser heating. The formation of diamond is possible even under the conditions of a typical geotherm (i.e., pressures of 70 GPa or higher). This does not mean, however, that all of the diamonds that originated in the lower mantle were formed according to the process described in our model. Ferro-periclase, a major constituent mineral in the pyrolitic lower mantle, is occasionally observed as inclusions in diamonds. So far this has been used as evidence of the origin of diamond in the pyrolitic lower mantle (Stachel et al. 2000; Kaminsky et al. 2001; McCammon 2001; Liu 1999). As our model describes subducted carbonates surrounded by MORB composition rather than pyrolitic composition, alternative formation mechanisms might be required to explain the origin of diamonds that contain ferro-periclase inclusions (Liu 1999; McCammon 2006); Liu (1999) has proposed that the magnesite itself would break down to diamond-bearing assemblages through the reaction, MgCO3 = MgO + C + O2. McCammon (2006) has proposed that the iron-disproportionation reaction involving subducted carbonates, 4Fe2+O + MgCO3 = MgO + C + 2Fe3+ 2O3, would take place in the lower mantle. It is, however, not necessarily the case that all lower mantle diamonds have ferro-periclase inclusions. Our model described as Reactions 2 and 3 may be another possible mechanism for diamond formation in the lower mantle. To confirm this, it will be necessary to examine the origin of diamonds that contain inclusions of SiO2, MgSiO3, and MgCO3 in the context of Reactions 2 and 3.
References
Akahama Y, Kawamura H (2004) High-pressure Raman spectroscopy of diamond anvil to 250 Gpa: method for pressure determination in the multimegabar pressure range. J Appl Phys 96:3748–3751
Akahama Y, Kawamura H, Hausermann D, Hanfland M, Shimomura O (1995) New high-pressure structural transition of oxygen at 96 GPa associated with metallization in a molecular-solid. Phys Rev Lett 74:4690–4693
Andrault D, Angel RJ, Mosenfelder JL, Le Bihan T (2000) Equation of state of stishovite to lower mantle pressures. Am Mineral 88:301–307
Biellmann C, Gillet P, Guyot F, Peyronneau J, Reynard B (1993) Experimental evidence for carbonate stability in the Earth’s lower mantle. Earth Planet Sci Lett 118:31–41
Dasgupta R, Hirschmann MM, Dellas N (2005) The effect of composition on the solidus of carbonated eclogite from partial melting experiments at 3 GPa. Contrib Mineral Petrol 149:288–305
Fiquet G, Guyot F, Kunz M, Matas J, Andrault D, Hanfland M (2002) Structural refinements of magnesite at very high-pressure. Am Mineral 87:1261–1265
Hammouda T (2003) High-pressure melting of carbonated eclogite and experimental constraints on carbon recycling and storage in the mantle. Earth Planet Sci Lett 214:357–368
Hirose K, Fei Y, Ma Y, Mao H (1999) The fate of subducted basaltic crust in the Earth’s lower mantle. Nature 397:53–56
Hirose K, Takafuji N, Sata N, Ohishi Y (2005) Phase transition and density of subducted MORB crust in the lower mantle. Earth Planet Sci Lett 237:239–251
Iota V, Yoo CS (2001) Phase diagram of carbon dioxide: evidence for new associated phase. Phys Rev Lett 86:5922–5925
Isshiki M, Irifune T, Hirose K, Ono S, Ohishi Y, Watanuki T, Nishibori E, Takata M, Sakata M (2003) Stability of magnesite and its high-pressure form in the lowermost mantle. Nature 427:60–63
Kaminsky FV, Zakharchenko OD, Davies R, Griffin WL, Khachatryan-Blinova GK, Shiryaev A (2001) Superdeep diamonds from the Juina area, Mato Grosso State, Brazil. Contrib Mineral Petrol 140:734–753
Liu L-G (1999) Genesis of diamonds in the lower mantle. Contrib Mineral Petrol 134:170–173
Luth RW (2001) Experimental determination of the reaction aragonite + magnesite = dolomite at 5–9 GPa. Contrib Mineral Petrol 141:222–232
Morlidge M, Pawley A, Droop G (2006) Double carbonate breakdown reactions at high-pressures: an experimental study in the system CaO–MgO–FeO–MnO–CO2. Contrib Mineral Petrol 152:365–373
McCammon C (2001) Deep diamond mysteries. Science 293:813–814
McCammon C (2006) Microscopic properties to macroscopic behavior: the influence of iron electronic state. J Mineral Petrol Sci 101:130–144
Nishio-Hamane D, Nagai T, Fujino K, Seto Y, Takafuji N (2005) Fe3+ and Al solubilities in MgSiO3 perovskite: implication of the Fe3+AlO3 substitution in MgSiO3 perovskite at the lower mantle condition. Geophys Res Lett 32:L16306. doi :10.1029/2005GL023529
Occelli F, Loubeyre P, Letoullec R (2003) Properties of diamond under hydrostatic pressures up to 140 GPa. Nat Mater 2:151–154
Perterrmann M, Hirschmann MM (2003) Partial melting experiments on a MORB-like pyroxenite between 2 and 3 GPa: constraints on the presence of pyroxenite in basalt source regions from solidus location and melting rate. J Geophys Res 108(B2):10. doi :1029/2000JB000118
Ringwood AE, Irifune T (1988) Nature of the 650-km seismic discontinuity: implications for mantle dynamics and differentiation. Nature 331:131–136
Sano Y, Williams NS (1996) Fluxes of mantle and subducted carbon along convergent plate boundaries. Geophys Res Lett 23:2749–2752
Sato K, Katsura T (2001) Experimental investigation on dolomite dissociation into aragonite plus magnesite up to 8.5 Pa. Earth Planet Sci Lett 184:529–534
Shim S-H, Duffy TS (2000) Constraints on the P–V–T equation of state of MgSiO3 perovskite. Am Mineral 85:354–363
Stacey FD (1992) Physics of the Earth. (3rd edn.) Brookfield Press, Brisbane
Stachel T, Harris JW, Brey GP, Joswig W (2000) Kankan diamonds (Guinea) II: lower mantle inclusion parageneses. Contrib Mineral Petrol 140:16–27
Takafuji N, Nagai T, Fujino K, Seto Y, Hamane D (2006) Decarbonation reaction of magnesite in subducting slabs at the lower mantle. Phys Chem Mineral 33:651–654
Tschauner O, Mao H, Hemley R (2001) New transformations of CO2 at high-pressures and temperatures. Phys Rev Lett 87:75701-1–75701-4
Yaxley GM, Brey GP (2004) Phase relations of carbonate-bearing eclogite assemblages from 2.5–5.5 GPa: implications for petrogenesis of carbonatites. Contrib Mineral Petrol 146:606–619
Yoo CS, Cynn H, Gygi F, Galli G, Iota V, Nicol M, Carlson S, Hausermann D, Mailhiot C (1999) Crystal structure of carbon dioxide at high-pressure: “superhard” polymeric carbon dioxide. Phys Rev Lett 83:5527–5530
Acknowledgments
X-ray observations were conducted at SPring-8 (BL10XU, proposal No. 2006B1184, 2007A1510) and KEK-PF (BL13A, proposal No. 2005G143). This research was supported by a MEXT Grant-in-Aid for the 21st century COE Program on “Neo-Science of Natural History” at Hokkaido University, Japan. Y.S. and D.H. are also supported by JSPS research fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Seto, Y., Hamane, D., Nagai, T. et al. Fate of carbonates within oceanic plates subducted to the lower mantle, and a possible mechanism of diamond formation. Phys Chem Minerals 35, 223–229 (2008). https://doi.org/10.1007/s00269-008-0215-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-008-0215-9