
Abstract
High-pressure synthesis of a new SrSi2O5 phase was performed at 16 GPa and 900°C by using a Kawai-type multianvil apparatus. The powder X-ray diffraction pattern of the compound was analyzed by Rietveld refinement based on the structure of a high-pressure polymorph of BaGe2O5, BaGe2O5 III. The structure is orthorhombic with space group Cmca and cell parameters of a = 5.2389(1) Å, b = 9.2803(2) Å, c = 13.4406(1) Å, V=653.46(2) Å 3 (Z=8, ρcalc=4.549 g/cm3). The structure consists of layers containing SiO6 octahedra and SiO4 tetrahedra. In a unit layer, oxygen and strontium atoms are arranged in an approximation to hexagonal close-packing. The strontium atom is accommodated in a 12-coordinated site. Each SiO6 octahedron shares four corners with SiO4 tetrahedra and the other two corners with another SiO6 octahedra. The SiO6 octahedra are linked to each other to form SiO6 chains along the a-axis. This is the first known example of a silicate with a BaGe2O5 III-type structure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since Sr is an alkaline-earth metal element like Mg and Ca and has a slightly larger ionic radius than Ca, it is interesting to examine the substitution of Sr for Ca in Ca-based structures from the crystal chemistry point of view. Especially, strontium metasilicate, SrSiO3, is a potentially good analog of CaSiO3 which is one of the major endmember components in the constituent minerals of the Earth’s crust and mantle. The high-pressure phase relations of SrSiO3 were investigated by Shimizu et al. (1970) and Fleischer and DeVries (1988). In particular, Shimizu et al. (1970) reported the results of high-pressure experiments up to 12 GPa, based on the pressure scales of those days. According to their results, pseudowollastonite-type SrSiO3 (SrSiO3 I), which is stable at 1 atm, transforms to SrSiO3 II at about 3.5 GPa and to SrSiO3 III at about 6 GPa. The structure of SrSiO3 II was determined to be a walstromite-type structure and that of SrSiO3 III to contain four-membered tetrahedra rings (Machida et al. 1982). In our high-pressure experiments in the SrO–SiO2 system to research phase relations of SrSiO3 at higher pressure, it has been found that SrSiO3 III decomposes to SrSi2O5 plus Sr2SiO4 at about 11 GPa and 1000°C. This high-pressure decomposition behavior of SrSiO3 is similar to that of CaSiO3. The structure of the Sr2SiO4 phase was identified as being the Ca2SiO4 larnite-type, whereas the structure of the SrSi2O5 phase has not been previously determined.
In the case of CaSiO3, Kanzaki et al. (1991) first reported that CaSiO3 walstromite decomposes to CaSi2O5 + Ca2SiO4 at 10 GPa and 1f500°C and proposed the titanite (CaSiTiO5) structure type as a candidate of the CaSi2O5 structure. Angel et al. (1996) found that, after pressure release to ambient conditions, the CaSi2O5 phase has a triclinic structure derived from that of titanite. A feature specific to the triclinic phase of CaSi2O5 is that it contains fivefold-coordinated silicon sites in addition to four- and six-coordinated silicon sites. Furthermore, it was shown by Angel (1997) that, under hydrostatic compression, the triclinic CaSi2O5 transforms to a monoclinic titanite phase which contains only four- and six-silicon sites.
Our powder X-ray diffraction (XRD) pattern of the SrSi2O5 phase recovered from high pressure was inconsistent not only with a titanite-type structure, but also with any known silicate mineral. From comparison of XRD profiles among inorganic compounds with the AB2X5 stoichiometry, we have found that the XRD profile of SrSi2O5 phase agreed very well with that of BaGe2O5 III, reported by Ozima et al. (1982). Therefore, we have performed a Rietveld analysis of the high-pressure form of SrSi2O5 by using the crystal structure data for BaGe2O5 III as a model.
Experimental methods
The starting material was prepared from a mixture of reagent grade SrCO3 and silicic acid (SiO2·11wt% H2O) in the molar ratio of 1:2. The mixture was compressed into a pellet and heated at 1250°C for 12 h in a furnace. The powder XRD pattern of the product could be indexed as a mixture of pseudowollastonite-type SrSiO3 and SiO2 cristobalite. The pellet was reground into powder in an agate mortar for 1 h. High-pressure and high-temperature synthesis was performed by using the Kawai-type multianvil apparatus at Gakushuin University. Tungsten carbide anvils with a truncated edge length of 5 mm were used. A MgO octahedron was used as a pressure medium. A cylindrical Pt heater was put into the octahedron with a LaCrO3 sleeve and endplugs as thermal insulator. Tantalum discs were put between the LaCrO3 endplugs and the sample to prevent reaction between them. Temperature was measured with a Pt/Pt–13%Rh thermocouple. A hot junction of the thermocouple was placed on the outer surface of central part of the heater. The starting material was put in the Pt heater, and kept at 16 GPa and 900°C for 1 h, then quenched isobarically. More details on the high-pressure technique employed in this study are given in Suzuki and Akaogi (1995). The composition of the recovered sample was confirmed by SEM-EDS analysis. A microscopic observation of the recovered polycrystalline sample showed that many crystals had a tabular shape (5–20 μm in diameter, 1–2 μm in thickness).
The recovered sample was crushed and ground into powder. The powdered sample was mounted on a nonreflective quartz plate using acetone. The powder XRD pattern was obtained by using a Rigaku RINT 2500V diffractometer with monochromated Cr Kα radiation (45 kV, 250 mA) at Gakushuin University. The step-scanning method was applied for data collection in a 2θ range of 10–140°. The step size and counting time were 0.02° and 20 s per step, respectively.
Rietveld analysis was made by using the RIETAN-2000 program (Izumi and Ikeda 2000). Peak profiles were fitted with the pseudo-Voigt function. The preferred orientation was corrected with the March–Dollase function (Dollase 1986). As the observed powder diffraction pattern included weak peaks of SiO2 stishovite at 2θ=45.6° and 70.7°, which probably originated from unreacted cristobalite in the starting material owing to the relatively low-reaction temperature, stishovite was included in the refinement as an impurity phase.
Results and discussion
Results of Rietveld analysis
The powder XRD data for SrSi2O5 are shown in Table 1. All of the observed peaks, except for those of stishovite, can be assigned to BaGe2O5 III-type SrSi2O5. The calculated d values are very consistent with the observed ones. All of their reflection indices conform to the extinction rule of the space group Cmca. The results of the Rietveld analysis are shown in Table 2 and Fig. 1. When atomic displacement factors for all oxygen sites were refined independently, only O2 gave a negative value. Therefore, the isotropic atomic displacement factor for O2 was fixed at zero. The weighted chi-squared value (1.05) indicates that the profile represented by refined parameters fits well with the observed XRD data. The uncertainty of each XRD data point σ, which was used in the χ 2w examination, was estimated by applying the equation σi = 1.33×n −1/2i , where ni is a count number at the i-th data point. The coefficient of 1.33 was based on the result of Rietveld refinement of Si, the XRD pattern of which was taken with the same experimental conditions as those for SrSi2O5.

Results of the Rietveld analysis of BaGe2O5 III-type SrSi2O5. Cross and solid line show observed and calculated X-ray diffraction patterns, respectively. Vertical bars indicate the positions of Bragg reflection for SrSi2O5 (upper) and SiO2 stishovite (lower). The plot at the bottom of this figure represents the difference between the observed and calculated patterns
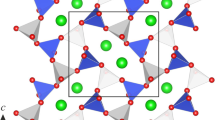
The refined BaGe2O5 III-type structure consists of silicon–oxygen framework layers which are perpendicular to the c-axis. In a unit layer, O and Sr are stacked to form a hexagonal close packing, where a layer of O1 and O3 is sandwiched between layers of O2, O4 and Sr (Fig. 2a). On the other hand, the stacking across the unit layers is regarded as a cubic close packing. Hence, a pattern of O and Sr packing in the unit cell can be described as ABACBC. Positions of Sr in the same layer are shown in Fig. 2b by the projection on (001). Si resides in an octahedral (Si1) and a tetrahedral (Si2) sites, and Sr is in a 12-fold coordination. Using the Si–O polyhedra as building blocks, two subunit layers can be constructed in the unit layer, as shown in Figs. 2a and 3. In the subunit layer, a SiO6 octahedron is connected by three corner-shared SiO4 tetrahedra. The SiO6 octahedra are linked via corners to form chains parallel to a-axis. The SrO12 polyhedra are positioned between the unit layers with linking each other by face sharing.

Projections of the crystal structure of BaGe2O5 III-type SrSi2O5 (a) along the a-axis and (b) along the c-axis. The rectangles indicate a unit cell dimension

Crystal structure of BaGe2O5 III-type SrSi2O5. Tetrahedra and octahedra show SiO4 and SiO6 polyhedra, respectively. Position of Sr is represented by sphere
Calculated interatomic distances and angles are listed in Table 3. The average Si1–O distance (1.797 Å) shows very good agreement with average Si–O distance of 1.796 Å for SiO6 octahedron among various high-pressure silicates (Finger and Hazen 2000). The details of the obtained SiO6 octahedron are described as follows. The Si1–O4 distance is the longest among the Si1–O distances (0.1 Å longer than the average Si1–O distance). On the other hand, Si1–O1 and Si1–O3 distances are about 0.1 Å shorter than that of the average Si1–O distance. Most of O–Si1–O angles are close to 90° which is the one for an ideal octahedron. Only O3–Si1–O3 angle indicates about 10° larger value than the others. This is caused by the off-center of Si1 on a quadrilateral plane consisting of O3 and O4. These descriptions indicate that the octahedra in SrSi2O5 phase are slightly distorted. In the tetrahedra, the average Si2–O distance (1.618 Å) is almost the same within the errors as the average Si–O distance of 1.611(8) Å in coesite (Geisinger et al. 1987), a typical high-pressure silicate consisting of SiO4 tetrahedra. However, the Si2–O1 distance is about 0.1 Å shorter than the others. The considerably large isotropic atomic displacement parameter for O1 suggests that an actual Si2–O1 distance might be longer than the value calculated by the average atomic positions. The bond valence sum (Brown and Altermatt 1985) of Si ion in the tetrahedron gives an acceptable value of 4.10 in spite of the unusually short Si2–O1 distance. The average Sr–O distance of 2.734 Å is comparable to that for cubic SrTiO3 perovskite (2.76 Å : Mitchell et al. 2000), which also contains 12-coordinated Sr. As corner-linked polyhedra in this structure are not allowed to tilt, it is suggested that the adjustment of size of the divalent cation site is attained to accommodate Sr2+ by those deformations of SiO4 tetrahedra and SiO6 octahedra. This adjustment results in the difference in distances between the stacking layers of oxygen and strontium. Namely, the distance between unit layers becomes larger than the thickness of a subunit layer. This difference causes the distortion of SrO12 polyhedra. The distortion results in a slight deviation of the Sr position from the mean level of O2 and O4 in the same layer. The distance between unit layers is about 33% longer than the thickness of a subunit layer. In BaGe2O5 III, the difference between them is about 27%. The ionic radius ratios are 3.60 for XIISr2+/VISi4+ and 2.96 for XIIBa2+/VIGe4+ using ionic radii by Shannon and Prewitt (1969). The comparison between the difference in the packing-layer thickness and the ionic ratios suggests that the relative ionic size of a divalent cation to that of a tetravalent cation may determine how much the thickness of the subunit layer should be shortened to adjust the space for the divalent cation.
Comparison with titanite type CaSi2O5
The titanite-type monoclinic CaSi2O5 comprises chains of corner-sharing SiO6 octahedra running parallel to the a-axis. SiO4 tetrahedra in the monoclinic CaSi2O5 connect the SiO6 octahedral chains with corner-sharing. BaGe2O5 III-type SrSi2O5 does not contain such chains but layers consisting of SiO6 octahedra and SiO4 tetrahedra as described above. In terms of a divalent cation environment, Ca in the monoclinic CaSi2O5 has a coordination number of seven and an average Ca-O distance of 2.363 Å (Angel 1997). On the other hand, Sr in SrSi2O5 is in a 12-fold site with the average Sr–O bond length of 2.734 Å . It seems to be possible that the divalent cation site in the titanite-type structure is adjusted to accommodate a larger ion than Ca2+ by tilting the SiO6 and SiO4 polyhedra. According to Hammonds et al. (1998), however, no rigid unit mode for the titanite structure indicates that it is difficult for the Si(Ti)–O polyhedra to tilt, where the rigid unit mode is a rotational vibration mode without deformation of the polyhedra. This means Si–O polyhedra have to be deformed by shortening Si–O distances to accommodate Sr2+ in the site without any tilting. Since the shortened Si–O bond distance results in an energetically less stable structure, BaGe2O5 III-type structure could be favorable for SrSi2O5 due to the larger divalent cation site than that in the titanite structure. The BaGe2O5 III-type structure can be a potential candidate for high-pressure silicate containing relatively large cations such as Ba. Furthermore, as each unit layer of tetravalent cation–oxygen polyhedra is isolated by layers consisting of only divalent cations, this type of crystal structure may be expected to have practical applications (e.g., as an ionic conductor).
References
Angel RJ (1997) Transformation of fivefold-coordinated silicon to octahedral silicon in calcium silicate, CaSi2O5. Am Mineral 82:836–839
Angel RJ, Ross NL, Seifert F, Fliervoet TF (1996) Structural characterization of pentacoordinate silicon in a calcium silicate. Nature 384:441–444
Brown ID, Altermatt D (1985) Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database. Acta Cryst B 41:244–247
Dollase WA (1986) Correction of intensities for preferred orientation in powder diffractometry: application of the March model. J Appl Cryst 19:267–272
Finger LW, Hazen RM (2000) Systematics of high-pressure silicate structures. Rev Mineral Geochem 41:123–155
Fleischer JF, DeVries RC (1988) Pressure–temperature diagram for the system SrSiO3. Mater Res Bull 23:609–612
Geisinger KL, Spackman MA, Gibbs GV (1987) Exploration of structure, electron density distribution, and bonding in coesite with Fourier and pseudoatom refinement methods using single-crystal X-ray diffraction data. J Phys Chem 91:3237–3244
Hammonds KD, Bosenick A, Dove MT, Heine V (1998) Rigid unit modes in crystal structures with octahedrally coordinated atoms. Am Mineral 83:476–479
Izumi F, Ikeda T (2000) A Rietveld-analysis program RIETAN-98 and its applications to zeolites. Mater Sci Forum 321–324:198–204
Kanzaki M, Stebbins JF, Xue X (1991) Characterization of quenched high pressure phases in CaSiO3 system by XRD and 29Si NMR. Geophys Res Lett 18:463–466
Machida K, Adachi G, Shiokawa J, Shimada M, Koizumi M, Suito K, Onodera A (1982) High-pressure synthesis, crystal structures, and luminescence properties of europium(II) metasilicate and europium(II)-activated calcium and strontium metasilicates. Inorg Chem 21:1512–1519
Mitchell RH, Chakhmouradian AR, Woodward PM (2000) Crystal chemistry of perovskite-type compounds in the tausonite-loparite series, (Sr1–2xNa x La x )TiO3. Phys Chem Minerals 27:583–589
Ozima M (1985) Structure of high-pressure phases of barium germanium oxide, BaGe2O5. Acta Cryst C 41:1003–1007
Ozima M, Susaki J, Akimoto S, Shimizu Y (1982) The system BaO–GeO2 at high pressures and temperatures, with special reference to high-pressure transformations in BaGeO3, BaGe2O5, and Ba2Ge5O12. J Sold State Chem 44:307–317
Shannon RD, Prewitt CT (1969) Effective ionic radii in oxides and fluorides. Acta Cryst B 25:925–946
Shimizu Y, Syono Y, Akimoto S (1970) High-pressure transformations in SrGeO3, SrSiO3, BaGeO3, and BaSiO3. High Temp High Press 2:113–120
Suzuki T, Akaogi M (1995) Element partitioning between olivine and silicate melt under high pressure. Phys Chem Mineral 22:411–418
Acknowledgements
We thank Drs. Y. Inaguma, T. Katsumata and T. Kuribayashi for their helpful advises on Rietveld analysis, and also Dr. Chakhmouradian and an anonymous reviewer for constructive reviews. This study was supported in part by Grants-in-Aid from the Ministry of Education, Science and Culture, Japan, and Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kojitani, H., Kido, M. & Akaogi, M. Rietveld analysis of a new high-pressure strontium silicate SrSi2O5. Phys Chem Minerals 32, 290–294 (2005). https://doi.org/10.1007/s00269-005-0467-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-005-0467-6