
Abstract
Background
Pancreatic neuroendocrine tumors (PNETs) are a characteristic feature of the tumor syndromes multiple endocrine neoplasia type 1 (MEN-1) and von Hippel-Lindau disease (VHL). With VHL, about 10% of the patients exhibit PNETs by age 40 years. Metastatic potential is high if the tumors have grown to >3 cm in diameter. Optimal surgical treatment is still a challenge.
Methods
We report three cases, all women, ages 22, 30, and 39 years, respectively, who had known VHL, confirmed by classic organ manifestations and germline mutations of the VHL gene. All were diagnosed, in an asymptomatic stage, with solid tumors of the pancreatic tail or tail/corpus area measuring 2.9–5.6 cm diameter. All accepted the offer of laparoscopic organ-sparing removal of the tumors.
Results
In all three cases, the tumor was entirely removed. In two cases, resection of the spleen was also necessary as dissection of the tumor from the major splenic vessels was impossible. Operating time was 215–365 min, and blood loss was 200–700 ml. Histolopathology revealed benign PNETs in two cases, but the third patient had regional lymph node metastases. There were no complications, and the hospital stay was 4–7 days.
Conclusions
Organ-sparing laparoscopic surgery is an important option for treating VHL-associated PNETs of the pancreatic tail.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Von Hippel-Lindau disease (VHL) is an autosomal dominant disorder with an incidence of about 1:36.000 [1]. VHL is caused by germline mutations of the VHL gene located on 3p25-6, and mutations are identified in virtually all families [1, 2]. VHL is characterized by tumors affecting various organs. Major manifestations are hemangioblastomas of the retina and the central nervous system (CNS), renal cell carcinoma, pheochromocytomas, endolymphic sac tumors, and tumors of the epididymis or broad ligament [1, 3, 4]. The pancreas is often altered by cystic or neoplastic changes. Pancreatic cysts are frequent and form serous cystadenomas in some patients. All of these cystic pancreatic lesions are benign and do not need treatment. In contrast, up to about 10% of VHL patients exhibit solid pancreatic tumors [1, 5, 6]. Histologically, these noncystic neoplasias are pancreatic neuroendocrine tumors (PNETs) [7]. PNETs can become malignant and develop metastases, and they may be life-threatening [5].
Surgical treatment of VHL-associated PNET is a major challenge. The reports so far available have described laparotomy with partial or complete resection of the pancreas, which is currently regarded as the state-of-the-art treatment. Here, for the first time, we report laparoscopic organ-sparing removal of PNETs in patients with VHL.
Methods
The German registry for VHL was established during the 1980s at the University Medical Center in Freiburg. All registrants were investigated and documented by a uniform program including molecular genetic analysis of the VHL gene and clinical screening that included retinoscopy and magnetic resonance imaging (MRI) of the CNS and abdomen for the entire spectrum of VHL. Since 2009, we have considered laparoscopic surgery as an optimal new method for surgery of VHL-associated PNETs. All patients with PNETs >3 cm diameter were considered for this treatment if the tumors were located in the pancreatic body or tail region.
Radiological imaging was performed by standard MRI and/or computed tomography (CT) with intravenously administered contrast medium. Notably, MRI contrast medium enhancement was studied during the early arterial phase. Molecular genetic analyses included direct sequencing of all exons of the VHL gene and multiplex ligation-dependent probe amplification (MLPA) testing for large rearrangements and deletions [2].
Results
Case 1
The 24 year-old woman was diagnosed at age 20 with VHL. She had bilateral retinal angiomas that were treated by laser coagulation in 2003. The left eye was treated by laser coagulation several times and subsequently by vitrectomy. Molecular genetic investigation of a blood sample showed a large deletion of exon 3 of the VHL gene. The family history was negative.
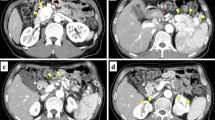
The patient was admitted for a complete clinical investigation to the Freiburg Preventive Medicine Center. MRI of the CNS revealed multiple hemangioblastomas of the spinal cord but no intracerebral hemangioblastomas. MRI of the abdomen showed multiple cysts in the pancreatic head with a maximum diameter of 4 cm and pancreatic duct ectasia. In addition, a lesion was detected in the pancreatic tail that measured 2.3 × 1.6 cm in the axial view (Fig. 1a). There was moderate contrast enhancement of the tumor. The kidneys and paraganglia were without pathological changes. Two years later, the tumor had grown to 1.9 × 2.9 cm.

Case 1. a Magnetic resonance imaging (MRI) of the upper abdomen with intravenous contrast medium shows a 2.9 cm diameter pancreatic neuroendocrine tumor (PNET) (arrows). b Histology shows a pancreatic endocrine neoplasm with a microtrabecular growth pattern; the tumor is circumscribed with pushing borders. At the upper left edge is a residual Langerhans endocrine islet (H&E)
Laparoscopic surgery was performed with five ports (two 10 mm, three 5 mm). The pancreatic tail was transected using a bipolar vessel sealing instrument (Ligasure; Covidien, Miami, FL, USA). Because the tumor had infiltrated the splenic vessels, splenectomy was necessary. Finally, the tissue was inserted in a bag. After morcellating the spleen in a bag, all tissue was removed through a 2.5-cm trocar site in the left upper abdomen. Operating time was 365 minutes, and estimated blood loss was 700 ml.
The postoperative course was uneventful. The patient was discharged home on day 6. Histology demonstrated a completely extirpated 3.5-cm benign PNET (Fig. 1b). The patient was reinvestigated 10 months after surgery. Abdominal MRI did not show any tumor in the remnant pancreas, adjacent lymph nodes, or liver. The cosmetic and clinical result was considered optimal by the patient. She returned to normal life about 6 weeks after surgery.
Case 2
A female patient observed reduced vision in the right eye at age 17 years. She was diagnosed with retinal angiomatosis of the right eye. Laser therapy and brachytherapy were performed. Vision of the right eye remained reduced to 15%. At age 24, a hemangioblastoma of the cervical spinal cord was resected. At age 27, she underwent craniotomy for a cerebellar hemangioblastoma. Genetic testing revealed the mutation VHL c.394 C > T, which predict a truncated VHL protein (Q132X). In 2007, a 20 mm diameter left renal tumor was detected but not resected owing to its small size, as internationally recommended [1].
In 2009 at age 38, abdominal MRI and CT angiography showed, in the axial view, a solid, 3.6 × 3.3 cm tumor of the pancreatic tail (Fig. 2a). Early arterial phase imaging after contrast injection revealed that the tumor had significant hyperperfusion. In addition, multiple pancreatic cysts were noted.

Case 2. a MRI of the upper abdomen with intravenous contrast medium shows a 3.6 cm diameter PNET (arrows). b Surgical specimen of a 3.2 cm diameter PNET. c Histology shows a pancreatic endocrine neoplasm with focal clear cell change at the right (H&E)
Four ports were inserted in the upper abdomen (two 10 mm, two 5 mm) for laparoscopic surgery. After dissecting the gastrocolic ligament, the lesser sac was opened. Following diversion of the splenocolic ligament, the pancreatic tail was explored. Because the tumor was localized in the low-lateral region, a spleen-preserving resection could be performed. A Ligasure was used for transecting the pancreas. Additionally, an 8-cm retrogastric cyst was fenestrated and drained. Finally, the tissue was inserted in a bag and removed through a 2.5-cm trocar site in the left upper abdomen. Operating time was 215 minutes, and estimated blood loss was 200 ml.
The postoperative course was uneventful except for a persistent fistula draining pancreatic fluid for 3 weeks. The patient went home on day 7 and had fully recovered at 7 weeks. Final pathology demonstrated a completely resected 3.2-cm benign PNET and a serous cystadenoma (Fig. 2b, c).
Case 3
A female patient presented with VHL disease at age 16, when a symptomatic left adrenal pheochromocytoma was diagnosed and removed laparoscopically. At age 28, a cystic cerebellar hemangioblastoma was removed. Molecular genetic testing revealed the mutation VHL c.500 G > A (p. R167Q).
In 2010, she was admitted to the Freiburg Preventive Medicine Center. Baseline MRI detected multiple intraspinal hemangioblastomas associated with tumor cysts. Abdominal MRI also showed a 5.6 × 5.4 cm solid tumor in the pancreatic tail that was richly vascularized (Fig. 3a).

Case 3. a MRI of the upper abdomen with intravenous contrast medium shows a 5.6 cm diameter PNET (arrows). b Histology of a PNET metastasis (H&E)
Four ports (two 10 mm, two 5 mm) were inserted in the upper abdomen for laparoscopic surgery. Following mobilization of the left colonic flexure and widely opening the lesser sac by dissecting the short gastric vessels, the pancreatic tail was explored. There were dense adhesions of the tumor tissue to the splenic vein, so resection of the pancreatic tail and the spleen were indicated. The excised tissue was inserted in a bag and morcellated, after which the tissue was removed through a 3-cm trocar site in the left upper abdomen. Operating time was 360 minutes.
The patient was discharged from the hospital on day 4. Histopathologic examination demonstrated a completely resected PNET (measuring 5 cm). Three of eleven lymph nodes showed metastases (Fig. 3b).
Discussion
Pancreatic islet cell tumors occur mainly as sporadic entities. A significant subset of these tumors, however, is hereditable. Hereditary forms include multiple endocrine neoplasia type 1 (MEN-1) caused by germline mutations of the MEN1 gene and VHL caused by germline mutations of the VHL gene. In contrast to sporadic tumors, striking characteristics of hereditary tumors, in general, are multifocal occurrence and manifestation at a young age. VHL PNETs are virtually always nonfunctioning, as was described for all three of our reported patients. Recommendations drawn from clinical observation of various series of patients with VHL-associated PNETs showed that the likelihood for metastases is correlated with the diameter of the largest PNET if several tumors are detected. The critical size is regarded to be 3 cm diameter [8]. Therefore, it would be important to be able to resect these tumors before they are 3 cm, remembering that most of the time PNETs are asymptomatic. Patients with VHL represent an important group in which treatment of these tumors is highly challenging.
Here, we reported three cases in which PNETs were removed laparoscopically. The tumor sizes were 2.9, 3.6, and 5.6 cm (largest diameter), respectively. In all cases, the tumor was located in the pancreatic body and/or tail area and was successfully completely removed. Owing to impossible dissection from the spleen in two cases, splenectomy was also necessary. No patient experienced complications. No blood transfusion was needed. Postoperatively, there was no pancreatitis, and pain was well controlled by low doses of analgesics. All patients were discharged prior to a maximum 7 days in hospital. There were scars for up to five incisions of ≤2.5 cm length; one was in periumbilical tissue. All three of the women expressed their full satisfaction with the cosmetic result. In one case, lymph node metastases were shown after complete lymphadenectomy of the upper abdomen.
Von Hippel-Lindau disease is important in regard to organ-sparing surgery because of the different component tumors and the high likelihood of multifocal synchronous and metachronous disease. This surgery is now standard for renal cell carcinoma associated with VHL [9]. Organ-sparing surgery was introduced in 1999 for removal of adrenal pheochromocytomas primarily as an open operation. Subsequently, adrenal-sparing surgery has been combined with laparoscopic surgery for VHL-associated pheochromocytomas. Meanwhile, this technique has become state of the art for pheochromocytomas/paragangliomas [10].
The VHL-associated PNETs can be located in any part of the pancreas. Often, these tumors are incidentally detected, as in our cases, because most are asymptomatic. The patients who do not have any PNET-related symptoms are confronted with a surprising situation. Importantly, these patients are relatively young and run a high risk of developing multifocal tumors in various organs, mainly the eyes, CNS, kidneys, and adrenals. Thus, a minimally traumatic procedure to remove PNETs is mandatory.
Laparoscopic resection of sporadic PNETs has been reported by several groups as case reports or series [11–17]. However, the current reported cases are, to the best of our knowledge, the first descriptions of organ-sparing pancreatic surgery in VHL patients. This is of particular relevance as the patients had considerable clinical and cosmetic benefit in the setting of a disease affecting several organs that would eventually necessitate multiple resections. This technology has been proved useful and safe for patients with VHL-associated PNETs of the tail/corpus area of the pancreas. It may also be an encouraging option for a broad range of heritable neoplastic disorders of the pancreas.
References
Lonser RR, Glenn GM, Walther M et al (2003) von Hippel-Lindau disease. Lancet 361:2059–2067
Franke G, Bausch B, Hoffmann MM et al (2009) Alu–Alu recombination underlies the vast majority of large VHL germline deletions: molecular characterization and genotype–phenotype correlations in VHL patients. Hum Mutat 30:776–786
Maher ER, Nathanson K, Komminoth P (2004) Von Hippel-Lindau syndrome (VHL). In: DeLellis RA, Lloyd RV, Heitz PU et al (eds) World Health Organization classification of tumours of endocrine organs. IARC Press, Lyon, pp 230–237
Plate KH, Vortmeyer AO, Zagzag D (2007) Von Hippel-Lindau disease and haemangioblastoma. In: Louis DN, Ohgaki H, Wiestler OD et al (eds) WHO classification of tumours of the central nervous system. IARC Press, Lyon, pp 215–217
Corcos O, Couvelard A, Giraud S et al (2008) Endocrine pancreatic tumors in von Hippel-Lindau disease: clinical, histological, and genetic features. Pancreas 37:85–93
Woodward ER, Maher ER (2006) Von Hippel-Lindau disease and endocrine tumour susceptibility. Endocr Relat Cancer 13:415–425
Heitz P, Komminoth P, Perren A et al (2004) Pancreatic endocrine tumours. In: DeLellis RA, Lloyd RV, Heitz PU et al (eds) World Health Organization classification of tumours: pathology and genetics. Tumours of endocrine organs. IARC Press, Lyon, pp 175–182
Libutti SK, Choyke PL, Alexander HR et al (2000) Clinical and genetic analysis of patients with pancreatic neuroendocrine tumors associated with von Hippel-Lindau disease. Surgery 128:1022–1027 discussion 1027–1028
Grubb RL 3rd, Choyke PL, Pinto PA et al (2005) Management of von Hippel-Lindau-associated kidney cancer. Nat Clin Pract Urol 2:248–255
Neumann HP, Eng C (2009) The approach to the patient with paraganglioma. J Clin Endocrinol Metab 94:2677–2683
Kim SC, Park KT, Hwang JW et al (2008) Comparative analysis of clinical outcomes for laparoscopic distal pancreatic resection and open distal pancreatic resection at a single institution. Surg Endosc 22:2261–2268
Langer P, Fendrich V, Bartsch DK (2009) Minimally invasive resection of neuroendocrine pancreatic tumors. Chirurg 80:105–112
Assalia A, Gagner M (2004) Laparoscopic pancreatic surgery for islet cell tumors of the pancreas. World J Surg 28:1239–1247
Fernandez-Cruz L, Blanco L, Cosa R et al (2008) Is laparoscopic resection adequate in patients with neuroendocrine pancreatic tumors? World J Surg 32:904–917
Gagner M, Pomp A (1997) Laparoscopic pancreatic resection: is it worthwhile? J Gastrointest Surg 1:20–25 discussion 25–26
Mabrut JY, Fernandez-Cruz L, Azagra JS et al (2005) Laparoscopic pancreatic resection: results of a multicenter European study of 127 patients. Surgery 137:597–605
Spitz JD, Lilly MC, Tetik C et al (2000) Ultrasound-guided laparoscopic resection of pancreatic islet cell tumors. Surg Laparosc Endosc Percutan Tech 10:168–173
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
von Dücker, L., Walz, M.K., Voss, C. et al. Laparoscopic Organ-Sparing Resection of Von Hippel-Lindau Disease-Associated Pancreatic Neuroendocrine Tumors. World J Surg 35, 563–567 (2011). https://doi.org/10.1007/s00268-010-0878-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00268-010-0878-5