
Abstract
Mammalian cells are frequently at risk of DNA damage from multiple sources. Accordingly, cells have evolved the DNA damage response (DDR) pathways to monitor the integrity of their genome. Conceptually, DDR pathways contain three major components (some with overlapping functions): sensors, signal transducers, and effectors. At the level of sensors, ATM (ataxia telangiectasia mutated) and ATR (ATM-Rad3-related) are proximal kinases that act as the core sensors of and are central to the entire DDR. These two kinases function to detect various forms of damaged DNA and trigger DNA damage response cascades. If cells harbor DDR defects and fail to repair the damaged DNA, it would cause genomic instability and, as a result, lead to cellular transformation. Indeed, deficiencies of DDR frequently occur in human cancers. Interestingly, this property of cancer also provides a great opportunity for cancer therapy. For example, by using a synthetic lethality model to search for the effective drugs, ChK1 inhibitors have been shown to selectively target the tumor cells with p53 mutations. In addition, the inhibitors of poly(ADP-ribose) polymerase (PARP-1) showed selectively killing effects on the cells with defects of homologous recombination (HR), particularly in the context of BRCA1/2 mutations. Since Brit1 is a key regulator in DDR and HR repair, we believe that we can develop a similar strategy to target cancers with Brit1 deficiency. Currently, we are conducting a high-throughput screening to identify novel compounds that specifically target the Brit1-deficient cancer which will lead to development of effective personalized drugs to cure cancer in clinic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The replication of mammalian cells is a high-fidelity process that assures an accurate passage of genomic information to the daughter cells. However, their genome is constantly challenged by endogenous metabolic byproducts and environmental factors that can alter its chemical structure, corrupt its encoded message, and, as a result, lead to the improper presence of single-strand DNA breaks (SSBs) and/or double-strand DNA breaks (DSBs). Many cellular pathways have evolved to respond to these challenges (DNA damage response, DDR) to maintain genomic integrity in the host. The DNA damage response involves the sensing of DNA damage followed by transduction of the damage signal to a network of cellular pathways, including cell cycle checkpoints, DNA repair, and the apoptotic pathway.
Among all the types of damage, DSBs pose the greatest challenge to cells and are dangerous and potentially lethal lesions [1]. DNA-damaging agents such as ionizing radiation, reactive oxygen species, replication fork collapses, and dysfunctional telomeres can cause DSBs [2]. For a cell to preserve genome integrity, DSBs need to be repaired promptly, and the repair can be done through two independent but not mutually exclusive mechanisms initiated by DDR: error-prone nonhomologous end joining (NHEJ) or relatively error-free homologous recombination (HR) [1]. Efficient detection and appropriate repair of the damaged DNA are particularly important for dividing cells where replication or segregation of chromosomes bearing unrepaired lesions could seriously compromise genome integrity and eventually lead to DDR-related diseases such as cancer. In this review we introduce the major pathways of the DDR, describe the relationship of the DDR with cancer, and also discuss the novel approach of ongoing research on drug discovery to target cancer using the concept of synthetic lethality.
The major pathways for DDR
In general, the DDR network consists of two major parallel pathways that respond to different DNA damage, and each pathway is arbitrarily composed of sensors, transducers, and effectors [3–6]. In this network, two phosphatidylinositol-3-related kinases, ATM (ataxia telangiectasia mutated) and ATR (ATM-Rad3-related), are located at the top of checkpoint signal cascades, which phosphorylate and activate a variety of molecules to execute the DNA damage response (Fig. 1) [3–6]. ATM is activated primarily by DSBs induced by ionizing irradiation and acts during all phases of the cell cycle, whereas the ATR pathway responds to agents interfering with functions of DNA replication forks, such as ultraviolet light and hydroxyurea [3, 7]. ATM can also activate many of the downstream targets of the ATR pathway. In fact, some agents have been shown to be able to activate both pathways, suggesting that the two major pathways of the DDR are interlaced in a cell.

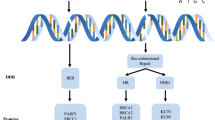
Brit1 is involved in regulation of ATM/ATR-dependent DNA damage response pathways. The DDR network consists mainly of two interconnected pathways, ATM and ATR. Responding to the DNA damage, these two core kinases ATM and ATR in the DDR are activated and recruit the sensors (such as the MRN complex, 53BP1, and MDC1 in the ATM pathway and RPA in the ATR pathway), transducers, and effectors proteins, and finally regulate the cell cycle checkpoint, DNA repair, and apoptosis. Brit1 protein participates in the two pathways and modulates the checkpoint and DNA repair through BRCA1 and ChK1 (shown as a black bold line)
At the level of sensors, ATM and ATR are proximal kinases that act as the core sensors of and are central to the entire DDR. These two kinases, by collaborating with other sensor molecules, function to detect various forms of damaged DNA and trigger DNA damage response cascades. For example, replication protein A (RPA, an ssDNA-binding protein) is required for recruitment of ATR and its partner protein ATRIP to the sites of DNA damage and also for ATR-mediated ChK1 activation [8]. Full activation of the DDR pathway induced by ATR also requires the Rad9-Rad1-HUS1 and Rad17-RFC complexes. It is possible that Rad17-RFC binds to the damage site and then loads the Rad9-Rad1-HUS1 complex, which could act as a scaffold so ATR can phosphorylate and activate its substrates [9]. Regarding the ATM pathway, the MRE11-Rad50-NBS1 (MRN) complex is a major sensor of broken DNA that recruits ATM to the break sites [10–12]. In addition, several other proteins such as 53BP1 [13, 14], MDC1 [15, 16], and Brit1/MCPH1 (hereafter Brit1) [17] might participate in this process to locate ATM or facilitate activation of ATM at the lesion sites of the DNA. Indeed, our recent studies demonstrated that Brit1 colocalized with all these sensor molecules and was required for their activation, suggesting that Brit1 may be positioned on the very top of ATM/ATR pathway (Fig. 1) [17].
At the level of transducers, ChK1 and ChK2 form the core module and lie downstream of and are mainly phosphorylated by ATM and/or ATR. These two kinases transduce signals from the sensors of the pathway flow to the effectors [6]. For instance, ChK1 is phosphorylated by ATM/ATR at Ser345 and Ser317 in response to various types of DNA damage which can be induced by ionizing radiation, ultraviolet light, hydoxyurea, and topoisomerase inhibitors [18–23]. ChK1, in turn, phosphorylates Ser123 and several other serine residues of the phosphatase CDC25A [21, 24], which target it for ubiquitin-dependent degradation [25]. As a result, CDC25A can no longer dephosphorylate and activate CDK2 and CDK1, causing cells to be arrested in late G1, S, and G2 phases [25, 26]. The role of ChK1 in DDR-induced checkpoints also has been clearly demonstrated using various ChK1 knockdown/knockout models [6, 19, 21, 22, 24, 27]. Thus, ChK1 is suggested to be one of the most important checkpoint proteins downstream of ATM/ATR. In parallel, ChK2 is a key kinase downstream of ATM and is primarily involved in DDR-induced apoptosis [6].
The effectors of the DDR include proteins that participate in DNA repair, transcription regulation, replication, cell-cycle control, and apoptosis; these proteins include CDC25, p53, and different DNA repair enzymes [4, 8]. Most of these effectors are downstream molecules of either ATM/ATR and/or ChK1/ChK2. Among them, p53 has a pivotal role in arresting cells in execution of the DDR [28]. p53 can be phosphorylated and stabilized by ATM/ATR and by another PIKK family member, DNA-PK. p53 can also be phosphorylated by ChK2, which itself functions as a substrate of ATM in response to DNA damage [5, 29]. The phosporylation of p53 by these DDR kinases leads to reduced binding of p53 to MDM2, which in turn allows replacement of ubiquitin moieties by acetylation, resulting in p53 stabilization and full activation [30]. Upon activation, depending on the severity of the stress signals, the pattern of post-transcriptional modifications, and the cellular context, p53, acting as a potent transcriptional activator and/or repressor, can induce cell-cycle arrest and/or apoptosis [28, 31–34], and remarkably p53 has also been shown to localize directly to sites of DNA damage and promote proper repair [35].
Tumorigenesis and DDR pathways
As described above, mammalian cells are frequently at risk of DNA damage from multiple sources, including ultraviolet light, ionizing radiation, and reactive oxygen species, and accordingly cells evolve the DDR pathways to monitor the integrity of their genome and repair any damaged DNA [36]. Indeed, intact DDR pathways are very critical for preventing the replication of damaged DNA templates and transmission of mutations to daughter cells. Therefore, defects in DDR will result in accumulation of genetic mutations, gene amplification, and chromosomal alterations, which in turn contribute to malignant transformation and tumorigenesis.
The key roles of the DDR pathways in tumor suppression have been well demonstrated by different research approaches. The first evidence is seen in patients with genetic instability syndromes. For instance, at the sensory level, human syndromes such as ataxia telangiectasia (AT) and Nijmegen breakage syndrome (NBS) results from gene deletion/mutation of the DDR pathway (herein are the genes ATM and NBS, respectively), and show defective responses to DSBs, striking effects in the nervous and immune systems, and cancer predisposition [37]. At the effector level of the DDR, cancer predisposition can be caused in many syndromes, including Bloom syndrome (defects in DNA helicases), Fanconi anemia (defects in the repair of DNA crosslinks), xeroderma pigmentosum (defects in the NER), Cockayne syndrome (defects in the NER), trichothiodystrophy (defects in the NER), and Li-Fraumeni syndrome (p53, tumor protein p53, TP53, Li-Fraumeni syndrome 1) [36, 38–41]. Li-Fraumeni syndrome has been studied for nearly three decades and is one of most studied syndromes in this category. This syndrome is due to a mutation in the p53 gene that greatly increases the susceptibility of patients to cancer [42, 43].
Second, many of the genes/proteins in the DDR are usually mutated and/or modified in somatic cancer tissues. For example, initiation of human breast, colon, lung, and urinary bladder tumors is characterized by phosphorylation of many proteins in the DDR, including the kinases ATM and ChK2, the histone H2AX, and one of the master regulators of cell cycle, p53 [44, 45]. As for genetic aberrations, it has been well known that patients who have loss-of-function mutations in BRCA1/2 genes are predisposed to breast and ovarian cancer [46]. Our studies on Brit1 also identified aberrations of Brit1 gene in several cancer lineages that link its deficiency to cancer initiation and progression [17]. In addition, epigenetic silencing of DDR genes has also been associated with tumorigenicity. For example, the promoters of BRCA1, MLH1, MGMT, and the helicase WRN have all been shown to be aberrantly methylated in human cancers [47]. In fact, the aberrations of DDR genes in cancer cells not only facilitate the initiation and progression of cancer but may also cause the cells’ resistance to many conventional cancer treatments. It is believed that cancer cells (and precancerous cells) generally grow under the condition of an increased level of endogenous DNA damage than do noncancer cells [44, 48, 49]. To survive in such a hostile environment, cancer cells must evolve new mechanisms to counteract the severe endogenous and exogenous genotoxic stress. This adaptation by cancer cells may indeed be responsible for their resistance to cancer treatment such as ionizing radiation and cytotoxic chemotherapy.
Finally, using gene knockout/knockdown technology, many studies have demonstrated that impairment of individual molecules in DDR such as p53 [50], 53BP1 [51], MDC1 [15], ATM [52], H2AX [53], and BRCA1 [54] can result in or further facilitate tumorigenesis. Taken together, deficiency of the DDR pathways clearly plays a crucial role in tumorigenesis and their response to treatment. By studying the DDR network and understanding how its deficiency is implicated in cancer, we may be able to develop drugs that can facilitate the DDR in normal cells to prevent cancer and the therapeutic agents that may specifically target DDR-deficient cancer for cancer treatment.
Drug development based on the deficiency of DDR: synthetic lethality model
Although it has been implicated in initiation of most cancers, DDR deficiency in cancer cells was also exploited therapeutically, such as radiotherapy and chemotherapeutics using DDR-targeting drugs. For example, the drugs that pharmacologically inhibit DNA repair have been proposed to be used to enhance chemosensitivity and radiosensitivity [55]. Recently, data indicate that deficiency in DNA repair actually acts as a therapeutic target in cancer [56]. Among the components of the DDR, the molecules with enzymatic activities were usually selected as a specific target of small molecule inhibitors, some of which are being evaluated in phase I/II studies [56]. These key enzymes include the PIKK family kinases (ATM/ATR and DNA-PK), ChK1/2, and PARP-1. Here we take a close look at ChK1/2 and PARP-1, both of which are suggestive of the synthetic lethality model, to search for the targets from DDR deficiency.
Currently, most therapies target proliferating cells rather than cancer cells per se. This can result in inefficient killing of cancer cells that have a low proliferative index. Furthermore, highly proliferative normal cells, such as cells in the bone marrow or gut, also are eradicated by the same therapies, which can reduce the patient’s quality of life and promote cancer recurrence due to immune suppression. Recently, however, the concept of synthetic lethality emerged. This concept was borrowed from classical genetics by Lee Hartwell and Stephen Friend to describe situations in which a mutation of gene A and a drug targeting gene B together cause a cell’s death [57]. This idea of synthetic lethality indeed has being used to develop novel drugs in cancer treatment. For example, ChK1, an important downstream kinase of the PIKKs, has recently been identified as the target of the staurosporine analog UCN-01, which causes abrogation of the G2/M checkpoint in response to ionizing radiation in p53-negative cells [58]. Thus, targeting the ChK1-related S and G2/M checkpoint activity would possibly lead to preferential sensitization of p53 mutant cancer cells to genotoxic agents. Based on this rationale, three agents targeting ChK1 (AZD7762, PF47736, and XL844) combined with gemcitabine are being evaluated in phase I studies, and it has also recently been disclosed that a phase I study of the Lilly/ICOS agent IC83 in combination with Pemetrexed has begun. Interestingly, AZD7762, PF47736, and XL844 also show a range of potencies against ChK2. Because ChK2 activates the G1 checkpoint and apoptotic pathways, it has been hypothesized that ChK2 inhibition may lead to enhanced effects of sensitizing p53-null cancer cells while protecting normal cells [59]. Therefore, ChK1/2 inhibitors (i.e., targeting the B gene in the above synthetic lethality model) might be suitable to selectively treat tumors with a p53-mutant background (i.e., the A gene mutation in the above model), which occurs in almost half of the malignant tumors.
Another example of synthetic lethality in cancer treatment are the inhibitors of poly(ADP-ribose) polymerase (PARP-1). PARP-1 is a key regulator of the base excision repair (BER) process and detects SSBs that arise from reactive oxygen species generated through normal and pathogenic cellular metabolic processes [60]. PARP-1 catalyzes ADP-ribose generated from nicotinamide adenine dinucleotide (NAD) to its downstream targets with a covalently charged branched chain of poly(ADP-ribose), which is known as poly(ADP-ribosyl)ation. The main target of this modification is believed to be PARP-1 itself, and the modified PARP-1 may recruit other components of the BER pathway to repair SSBs. Other potential targets of poly(ADP-ribosyl)ation include histones, transcription factors, and other signaling molecules such as NF-kB, DNA-PK, p53, topoisomerase I, and laminin B, which mainly couple DNA repair with regulation of the cell cycle [61].
Recently, some small molecules based on nicotinamide analogs are reported to function as inhibitors of PARP-1 [62, 63]. Notably, the emerging evidence indicates that PARP-1 inhibitors can sensitize tumor cells to cytotoxic therapies such as temozolomide, topoisomerase I inhibitors, platinum, and radiation [64–69]. In fact, a number of these molecules are being developed as clinical candidates to treat cancer. For example, AG014699 has been evaluated in the phase II study of human solid tumors administered temozolomide [70, 71]. Interestingly, inhibition of PARP-1 was found to selectively kill BRCA1- and BRCA2-deficient cells with minimal effect on wild-type cells and cells with heterozygosis of either BRCA1 or BRCA2 mutations [72, 73]. Further studies indicated that this selectivity of PARP-1 inhibitors was based on the defects of HR in cells [69]. Therefore, the inhibitors of PARP-1 may function as novel drugs to treat tumors with HR deficiency (Fig. 2). A clinical trial of one of the PARP-1 inhibitors (KU0059436) has begun in order to evaluate its pharmacokinetics and pharmacodynamics for the treatment of BRCA-associated cancer [74]; the trial may be expanded to treat all HR-deficient cancers in the future. PARP-1 inhibition is another example of using synthetic lethality in cancer treatment where HR genes represent gene A, which are defective in cancer, and PARP-1 inhibitors target the PARP-1 gene, the B gene (Fig. 2).

PARP-1 inhibitors as drugs for targeting Brca1-null cancer cells using the synthetic lethality concept. The inhibitors of PARP-1 inhibit mainly the BER pathway of the DDR (i.e., the SSB-related pathway, which is just like the B gene in the synthetic lethality model) and selectively kill the cells with defects in the components of HR (here just like the A gene mutation). Therefore, BRCA1/2-deficient cells with HR defects will be killed by PARP-1 inhibitors, whereas the normal cells with HR can survive
In addition to the examples described above, many new drugs can be developed to target cancer based on the concept of synthetic lethality, particularly in the context of DDR deficiency. Since virtually all cancers suffer from deficiency of certain DDR gene(s) or pathway(s) (i.e., A), we should be able to screen for drugs that target the complementary B pathway to achieve the specific killing. These drugs should have low toxicity to the normal cells because of the intactness of the A gene or pathway in the normal cells [75]. Because Brit1 deficiency has been shown in tumor-prone mice (K. Li, unpublished) and its aberrations have been significantly implicated in human cancer development [17], we currently are conducting a high-throughput screen to identify new compounds that can specifically target cancer with Brit1 deficiency. Our drug-screening program will certainly be expanded in the future to target the cancer with other DDR deficiencies with the ultimate goal of developing the most effective personalized drugs to cure cancer in clinic.
References
Bassing CH, Alt FW (2004) The cellular response to general and programmed DNA double strand breaks. DNA Repair 3:781–796
Featherstone C, Jackson SP (1999) DNA double-strand break repair. Curr Biol 9:R759–R761
Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15:2177–2196
Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3:155–168
Bakkenist CJ, Kastan MB (2004) Initiating cellular stress responses. Cell 118:9–17
Zhou BB, Bartek J (2004) Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer 4:216–225
Stokes MP, Rush J, Macneill J et al (2007) Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci USA 104:19855–19860
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300:1542–1548
Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297:547–551
D’Amours D, Jackson SP (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 3:317–327
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499–506
Petrini JH, Stracker TH (2003) The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol 13:458–462
Zgheib O, Huyen Y, DiTullio RA Jr et al (2005) ATM signaling and 53BP1. Radiother Oncol 76:119–122
Mochan TA, Venere M, DiTullio RA Jr et al (2004) 53BP1, an activator of ATM in response to DNA damage. DNA Repair 3:945–952
Lou Z, Minter-Dykhouse K, Franco S et al (2006) MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 21:187–200
Minter-Dykhouse K, Ward I, Huen MS et al (2008) Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J Cell Biol 181:727–735
Rai R, Dai H, Multani AS et al (2006) BRIT1 regulates early DNA damage response, chromosomal integrity, and cancer. Cancer Cell 10:145–157
Sanchez Y, Wong C, Thoma RS et al (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277:1497–1501
Liu Q, Guntuku S, Cui XS et al (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14:1448–1459
Zhao H, Piwnica-Worms H (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21:4129–4139
Zhao H, Watkins JL, Piwnica-Worms H (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 99:14795–14800
Gatei M, Sloper K, Sorensen C et al (2003) Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem 278:14806–14811
Xiao Z, Chen Z, Gunasekera AH et al (2003) Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem 278:21767–21773
Sørensen CS, Syljuåsen RG, Falck J et al (2003) Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3:247–258
Mailand N, Falck J, Lukas C et al (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science 288:1425–1429
Mailand N, Podtelejnikov AV, Groth A et al (2002) Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J 21:5911–5920
Zachos G, Rainey MD, Gillespie DA (2003) Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J 22:713–723
Rodier F, Campisi J, Bhaumik D (2007) Two faces of p53: aging and tumor suppression. Nucleic Acids Res 35:7475–7484
Pluquet O, Hainaut P (2001) Genotoxic and non-genotoxic pathways of p53 induction. Cancer Lett 174:1–15
Lavin MF, Gueven N (2006) The complexity of p53 stabilization and activation. Cell Death Differ 13:941–950
Marchenko ND, Zaika A, Moll UM (2000) Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 275:16202–16212
Murphy ME, Leu JI, George DL (2004) p53 moves to mitochondria: a turn on the path to apoptosis. Cell Cycle 3:836–839
Oren M (2003) Decision making by p53: life, death and cancer. Cell Death Differ 10:431–442
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116:205–219
Al Rashid ST, Dellaire G, Cuddihy A et al (2005) Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo. Cancer Res 65:10810–10821
Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366–374
McKinnon PJ, Caldecott KW (2007) DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet 8:37–55
van Brabant AJ, Stan R, Ellis NA (2001) DNA helicases, genomic instability, and human genetic disease. Annu Rev Genomics Hum Genet 1:409–459
D’Andrea AD (2003) The Fanconi road to cancer. Genes Dev 17:1933–1936
Cleaver JE (2005) Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer 5:564–573
Leibeling D, Laspe P, Emmert S (2006) Nucleotide excision repair and cancer. J Mol Histol 37:225–238
Malkin D, Li FP, Strong LC et al (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238
Srivastava S, Zou ZQ, Pirollo K et al (1990) Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348:747–749
Bartkova J, Horejsí Z, Koed K et al (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434:864–870
Di Micco R, Fumagalli M, Cicalese A et al (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444:638–642
Bertwistle D, Ashworth A (2000) BRCA1 and BRCA2. Curr Biol 10:R582
Jacinto FV, Esteller M (2007) Mutator pathways unleashed by epigenetic silencing in human cancer. Mutagenesis 22:247–253
Bartkova J, Bakkenist CJ, Rajpert-De Meyts E et al (2005) ATM activation in normal human tissues and testicular cancer. Cell Cycle 4:838–845
Gorgoulis VG, Vassiliou LV, Karakaidos P et al (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434:907–913
Donehower LA, Harvey M, Slagle BL et al (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356:215–221
Ward IM, Minn K, van Deursen J et al (2003) p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol 23:2556–2563
Lu S, Shen K, Wang Y et al (2006) Atm-haploinsufficiency enhances susceptibility to carcinogen-induced mammary tumors. Carcinogenesis 27:848–855
Bassing CH, Suh H, Ferguson DO et al (2003) Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114:359–370
Weaver Z, Montagna C, Xu X et al (2002) Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene 21:5097–5107
Ding J, Miao ZH, Meng LH et al (2006) Emerging cancer therapeutic opportunities target DNA-repair systems. Trends Pharmacol Sci 27:338–344
O’Connor MJ, Martin NM, Smith GC (2007) Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene 26:7816–7824
Garber K (2004) Running interference: pace picks up on synthetic lethality research. J Natl Cancer Inst 96:982–983
Busby EC, Leistritz DF, Abraham RT et al (2000) The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 60:2108–2112
Kawabe T (2004) G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 3:513–519
Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362:709–715
D’Amours D, Desnoyers S, D’Silva I et al (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342:249–268
Virág L, Szabó C (2002) The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54:375–429
Southan GJ, Szabó C (2003) Poly(ADP-ribose) polymerase inhibitors. Curr Med Chem 10:321–340
Calabrese CR, Almassy R, Barton S et al (2004) Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst 96:56–67
Miknyoczki SJ, Jones-Bolin S, Pritchard S et al (2003) Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther 2:371–382
Miknyoczki S, Chang H, Grobelny J et al (2007) The selective poly(ADP-ribose) polymerase-1(2) inhibitor, CEP-8983, increases the sensitivity of chemoresistant tumor cells to temozolomide and irinotecan but does not potentiate myelotoxicity. Mol Cancer Ther 6:2290–2302
Curtin NJ (2005) PARP inhibitors for cancer therapy. Expert Rev Mol Med 7:1–20
Ratnam K, Low JA (2007) Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res 13:1383–1388
Martin SA, Lord CJ, Ashworth A (2008) DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev 18:80–86
Plummer R (2005) First in human phase I trial of the PARP inhibitor AG-014699 with temozolomide (TMZ) in patients with advanced solid tumors. J Clin Oncol ASCO Ann Meet Proc 23:A3065
Plummer R (2006) First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma. J Clin Oncol ASCO Ann Meet Proc Part I 24:A8013
Bryant HE, Schultz N, Thomas HD et al (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913–917
Farmer H, McCabe N, Lord CJ et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917–921
Yap TA (2007) First in human phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of KU-0059436, a small molecule inhibitor of poly(ADP-ribose) polymerase (PARP) in cancer patients, including BRCA1/2 mutation carriers. J Clin Oncol ASCO Ann Meet Proc Part I 25:A3529
Kaelin WG Jr (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5:689–698
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liang, Y., Lin, SY., Brunicardi, F.C. et al. DNA Damage Response Pathways in Tumor Suppression and Cancer Treatment. World J Surg 33, 661–666 (2009). https://doi.org/10.1007/s00268-008-9840-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00268-008-9840-1