
Abstract
Purpose
We examined the effects of triptolide on receptor activator of nuclear factor-κB ligand (RANKL)-induced osteoclast differentiation and on titanium (Ti) particle-induced osteolysis.
Methods
To examine the effect of triptolide on osteoclast differentiation, bone marrow macrophages (BMMs) were treated with 100 ng/mL of RANKL and 30 ng/mL of macrophage-colony stimulating factor, or co-cultured with osteoblasts stimulated with 10 nM vitamin D3 and 1 μM prostaglandin E2 in the presence or absence of triptolide (2.8–14 nM). Osteoclast differentiation and activation were assessed using tartrate-resistant acid phosphatase staining, reverse transcriptase-polymerase chain reaction analysis to determine differentiation marker gene expression and pit formation assays. To examine the effect of triptolide on wear debris-induced osteolysis, titanium (Ti) particles were injected into the calvaria of ICR mice. Then, the mice were divided into three groups and were orally administered vehicle, or 16 or 32 μg/kg/day triptolide for ten days, followed by histomorphometric analysis.
Results
Triptolide suppressed RANKL-mediated osteoclast differentiation of BMMs in a dose-dependent manner. In a co-culture system, osteoblasts treated with triptolide could not induce osteoclast differentiation of BMMs, which was accompanied by down-regulation of RANKL and up-regulation of osteoprotegrin. Moreover, triptolide significantly inhibited bone resorption, and expression of the bone resorption marker genes. RANKL-induced activation of p38, ERK, and JNK was substantially inhibited by triptolide. Further, in a Ti-induced mouse calvarial erosion model, mice perorally administrated with triptolide showed significant attenuation of Ti-mediated osteolysis.
Conclusion
Our data indicated that triptolide had an anti-osteoclastic effect and significantly suppressed wear debris-induced osteolysis in mice.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
For centuries, Tripterygium wilfordii Hook F (TWHF), a traditional Chinese herb, has been used in treatment of autoimmune diseases such as rheumatoid arthritis (RA) [1, 2]. An active principal component of TWHF has been identified as triptolide, a diterpene triepoxide [3].
In addition to RA, triptolide has an array of biological activities, including anticancer [4] and immunosuppressive activities [5]. Anti-inflammatory and immunosuppressive activities of triptolide are mediated through inhibition of T cells, and inhibition of production of interleukin (IL)-1, IL-6, IL-8, tumour necrosis factor-α (TNF-α), and PGE2 by human peripheral blood monocytes [5]. In addition, triptolide has contraceptive and anti-cytogenesis effects [6, 7]. Despite its beneficial effects, triptolide has adverse effects and limitations such as cytotoxicity and poor water solubility [8–11].
To overcome the toxic effects and poor water solubility of triptolide, different triptolide derivatives have been synthesized [12–16]. In addition, efforts also have been made to identify beneficial effects of triptolide in other pathological conditions.
Periprosthetic osteolysis and subsequent loosening by wear particles of orthopaedic implants are the most common causes of total joint arthroplasty failure. Wear particles released from the prosthesis play a central role in initiation and development of osteolysis [17]. Macrophages engulfing wear particles such as Ti particles secrete high concentrations of pro-inflammatory cytokines including TNF-α, IL-6, and IL-1β. These factors directly and/or indirectly stimulate osteoclast formation and bone resorption [18–21]. In addition, periprosthetic osteolysis is initiated by activation of the receptor activator of nuclear factor-κB (RANK) and RANK ligand (RANKL) signaling pathways [22, 23], suggesting that osteoclasts are eventually recruited and activated, leading to osteolysis and consequent loosening of the prosthesis [24].
Osteoclasts are multinucleated cells derived from monocytes and macrophages [25]. RANKL and macrophage colony-stimulating factor (M-CSF), which are expressed in osteoblasts and stromal cells, induce differentiation of monocytes/macrophages into osteoclasts [26]. M-CSF regulates expression of RANK on the cell surface of monocytes/macrophages [27], which in turn responds to RANKL. Binding of RANKL and RANK on osteoclast progenitor cells evokes a cascade of signaling pathways, resulting in expression of genes involved in osteoclast differentiation and activation. Binding of RANKL to RANK triggers activation of tumor necrosis factor receptor-associated factor 6 (TRAF6) [28] and, subsequently, mitogen-activated protein kinases (MAPKs), such as extracellular signal-regulated kinase 1/2 (ERK1/2), p38, and stress activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) [29, 30]. RANKL/RANK signaling pathway leads to activation of transcription factors, such as nuclear factor of activated T cells (NFATc1), NF-κB, and c-Fos, and these transcription factors regulate expression of a series of osteoclast-specific genes, including cathepsin K (Cat K), tartrate-resistant acid phosphatase (TRAP), calcitonin receptor, and osteoclast-associated receptor [31, 32].
Since cells, secreted cytokines, and the signaling system involved in the activation of inflammatory cells and osteoclasts are similar, we investigated whether the anti-inflammatory compound triptolide could inhibit osteoclast differentiation and activity. In this study, we demonstrated the effects of triptolide on osteoclast differentiation and function, and investigated the underlying molecular mechanism. The inhibitory effect of triptolide on Ti-mediated inflammatory osteolysis in the calvaria of mice was also examined.
Materials and methods
Osteoclast differentiation
Male ICR mice (seven weeks old) were obtained from DBL Co. (Chungbuk, Korea). Bone marrow cells were isolated from the tibiae and femora of the mice, pooled, and cultured with α-MEM containing 10 % fetal bovine serum in a humidified incubator (5 % CO2 in air) at 37 °C. After culture for 24 hours, non-adherent cells were collected and centrifuged in a histopaque gradient centrifuge in order to obtain bone marrow mononuclear cells (BMM cells). For osteoclastogenesis, BMM cells were plated in a 96-well culture plate at a density of 2 × 104 cells/well, and cultured in α-MEM containing 100 ng/mL RANKL (Peprotech; London, UK) and 30 ng/mL M-CSF with or without 2.8, 7, or 14 nM of triptolide (Sigma-Aldrich; St. Louis, MO). For the co-culture system, mouse BMM cells were co-cultured with mouse calvarial osteoblastic cells for seven days in the presence of 10 nM vitamin D3 and 1 μM PGE2 (Sigma-Aldrich) [33].
Cell viability
The effect of triptolide on cell viability was determined using the methyl-thiazol tetrazolium (MTT) cytotoxicity assay (Sigma-Aldrich) as described previously [34]. For in vitro experiments, triptolide was dissolved in dimethyl sulfoxide (DMSO) to prepare a 500× stock solution. Control cells were treated with 0.2 % DMSO (vehicle). After treatment with indicated concentration of triptolide (2.8–14 nM) for two days, MTT was added to cultures. Insoluble formazan was extracted with dimethyl sulfoxide, and the absorbance of each well was measured at 490 nm using a 96-well microplate reader (Biorad; Hercules, CA).
TRAP staining and activity
TRAP staining was performed as described previously [33]. TRAP-positive multinucleated cells (MNCs) with three or more nuclei were scored. For measurement of TRAP activity, cells were fixed and then incubated with 100 μL of phosphatase substrate solution (3.7 mM p-nitrophenyl phosphate and 10 mM sodium tartrate in 50 mM citrate buffer, pH 4.6) at 37 °C for ten minutes. Following incubation, the enzyme reaction mixture was transferred to another plate and the reaction was stopped with 100 μL of 0.1 N NaOH. Absorbance was measured at 405 nm using an ELISA reader (BioRad; Hercules, CA).
Resorption pit formation assay
For the resorption pit assay, mouse BMM cells (2 × 104 cells/well) were seeded on bone slices (IDS; Boldon, UK) and treated with RANKL (100 ng/mL) and M-CSF (10 ng/mL) until multinucleated osteoclasts had formed. The cells were then incubated with RANKL in the presence or absence of 7 nM triptolide for an additional 48 hours. After the culturing period, osteoclasts were removed from bone slices by hematoxylin staining. The resorption pit area was measured using IMT i-solution (Daejeon, Korea).
RT-PCR
Total RNA was extracted from cells by using Tri-solution (Bioscience, Gyeongsan, Korea). First-strand complementary DNA (cDNA) was synthesized from 1 μg of total RNA by using the Super Script II First-Strand Synthesis System (Invitrogen). From the 20 μL cDNA reaction volume, 1–2 μL were used for each PCR assay. Primers and PCR conditions for Cat K, matrix metalloproteinase-9 (MMP-9) [35], c-Src [36], and integrin αv [37] were described previously [33]. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. cDNA was amplified by using rTaq polymerase (Takara, Tokyo, Japan) for a total of 25–35 cycles. PCR products were resolved by using agarose gel electrophoresis and images were captured with a gel documentation system. Real-time PCR reactions were performed with RANKL (F: 5′-GATTTTTCAAGCTCCGAGCTGG-3′ and R: 5′-CCTGAACTTTGAAAGCCCCAA-3′), and osteoprotegerin (OPG; F: 5′-ACTCGAACCTCACCACAGAGC-3′ and R: 5′-CGCTCGATTTGCAGGTCTTTC-3′) genes were performed with the SYBR® green PCR Master Mix (Applied Biosystems, Carlsbad, CA) using an ABI 7500 real-time PCR instrument (Applied Biosystems, USA). Each sample was analysed in triplicate and normalized to GAPDH expression. The expression levels were calculated as relative fold changes (the 2−∆∆Ct method was used).
Western blotting
Cells were lysed with RIPA buffer (50 mM Tris (pH 7.4), 1 % NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, 1 μg/mL pepstatin, and 1 μg/mL aprotinin), and the protein concentration was measured using a Bicinchoninic Acid Kit (Pierce; Rockford, IL). Cell lysates (20 μg) were separated on a SDS–polyacrylamide gel, and transferred to a PVDF membrane (Millipore; Billerica, MA) as described previously [38]. The membrane was blocked, incubated with primary antibodies and an appropriate secondary antibody coupled to horseradish peroxidase (UBI; Charlottesville, VA). The protein band was visualized by application of enhanced chemiluminescence reagents, and quantified with IMT i-Solution program.
Calvarial osteolysis model and histomorphometric analysis
Animal experiments were performed in accordance with principles and procedures approved by Kyungpook National University. A mouse calvarial osteolysis model in response to Ti particles was described previously [19]. Briefly, 15 healthy seven-week-old male ICR mice were randomly assigned to three groups, Ti particles with vehicle (Ti+Vehicle), Ti particles with triptolide (16 μg kg−1 day−1) (Ti+Tp 16), and Ti particles with triptolide (32 μg kg−1 day−1) (Ti+Tp 32). The mice were anaesthetized, the cranial periosteum was separated from the calvarium by sharp dissection, and 3 mg of Ti particles were placed directly on the surface of the bone, as described previously [19]. Triptolide stock solutions (1.8 and 3.6 mg/mL in DMSO) were diluted 1:370 with phosphate-buffered saline (PBS). Then, 100 μL of PBS containing vehicle (DMSO) or triptolide was orally administered to mice daily for ten days at doses of 16 and 32 μg/kg/day [39–43]. At the end of treatment, the mice were sacrificed, and the calvaria were excised, and fixed with 3.7 % formaldehyde in PBS (pH 7.4) for 48 hours. The specimens were washed with PBS and analysed using microcomputed tomography (μCT) (X-eye MCT; Seoul, Korea). After μCT imaging, the calvaria were decalcified in 10 % EDTA for one week, and histological sections were prepared, followed by staining with hematoxylin and eosin (H&E), and TRAP staining. The specimens were examined under a standard light microscope (Olympus, Japan) and photographed with a digital camera (Optical Systems, Germany). The lysed area was measured using IMT i-Solution.
Statistical analysis
Statistical analysis was performed with SPSS 21.0 (one-way analysis of variance; ANOVA) and p values of less than 0.05 or 0.01 were considered significant*(p < 0.05 and **p < 0.01).
Results
Triptolide inhibits osteoclast differentiation
To determine whether triptolide inhibits osteoclast differentiation, mouse BMM cells undergoing osteoclast differentiation in response to M-CSF and RANKL were treated with or without the indicated dose of triptolide. After four or five days, TRAP staining and activity were analyzed. As shown in Fig. 1, treatment with triptolide resulted in a significant dose-dependent reduction of both TRAP activity and TRAP-positive multinucleated cells. TRAP activity was significantly inhibited by 76.4 and 96 % at 7 and 14 nM triptolide, respectively. Similarly, the number of TRAP-positive multinucleated cells was also significantly decreased by 78.7 and 97.8 % at 7 and 14 nM triptolide, respectively. These results suggest that triptolide directly inhibits osteoclast differentiation. We also tested the effect of triptolide on osteoclast formation in a co-culture system comprising osteoblasts and BMM cells. Mouse calvarial osteoblasts and BMM cells were stimulated with vitamin D3 and PGE2 in the presence or absence of triptolide. As shown in Fig. 2, treatment with triptolide resulted in a significant reduction of TRAP activity and numbers of TRAP-positive osteoclasts (Fig. 2a and b). In addition, triptolide showed a tendency to inhibit the expression of RANKL in osteoblasts, and to induce the expression of osteoprotegrin (OPG), a decoy receptor for RANKL (Fig. 2c). These results suggest that triptolide may indirectly suppress osteoclast differentiation, targeting RANKL and OPG expression in osteoblasts. Collectively these results clearly demonstrate that triptolide directly and indirectly regulates osteoclast differentiation.

The inhibitory effect of triptolide on osteoclast formation of mouse bone marrow cells. a Mouse BMM cells were treated with M-CSF (30 ng/mL) and RANKL (100 ng/mL) for four or five days in the presence or absence of 2.8, 7, or 14 nM of triptolide. TRAP activity was measured at a wavelength of 405 nm, as described in the 'Materials and methods' section. (A, left panel) TRAP-positive MNCs containing three or more nuclei were scored and presented (A, right panel). bThe cells were stained with TRAP. The results are representative of three independent experiments. *p < 0.05 and **p < 0.01

The inhibitory effect of triptolide on osteoclast formation in a co-culture system. Mouse BMM cells and calvarial osteoblastic cells co-cultured for seven days in the presence of 10 nM vitamin D3 and 1 μM PGE2 were treated with the indicated concentration of triptolide. a TRAP activity and b TRAP-positive MNCs were measured, as shown in Fig. 1. b The cells were stained with TRAP. c Real time PCR for RANKL and OPG in calvarial osteoblasts stimulated with 10 nM vitamin D3 and 1 μM PGE2 for five days. The results are representative of three independent experiments
Triptolide inhibits osteoclast activity
To examine the effect of triptolide on bone resorption, osteoclasts cultured on bone slices were treated with or without triptolide. Analysis of resorbed surfaces of bone slices was performed using hematoxylin staining. As shown in Fig. 3, osteoclasts demonstrated good resorption of bone surface in the absence of triptolide, whereas bone resorption was dramatically inhibited by triptolide (7 nM; Fig. 3a). In addition, triptolide substantially inhibited the expression of genes associated with resorption, including integrin αv, Mmp-9, Cathespsin K, and c-Src (Fig. 3b). Although there are some limitations in densitometric analysis of RT-PCR, it clearly showed that the expression of genes associated with resorption was dramatically inhibited by triptolide. These results indicate that triptolide inhibits the resorption activity of osteoclasts.

Inhibition of bone resorption activity of osteoclasts by triptolide. Mouse BMM cells were seeded on dentine slices and treated with M-CSF (30 ng/mL) and RANKL (100 ng/mL) in order to induce differentiation into osteoclasts. Following formation of osteoclasts, the cells were treated with or without triptolide (7 nM) in the presence of M-CSF and RANKL for 48 h. a Resorption pit area was measured using an image analysis program. Scale bars =100 μm. b Changes in the expression of genes involved in osteoclast activity in response to triptolide (7 nM) were analyzed by using RT-PCR. The results are representative of three independent experiments
Triptolide inhibits the RANKL signaling pathway
To define the molecular mechanism(s) by which triptolide directly inhibits osteoclast differentiation and activation, phosphorylation of MAP kinases and activation of transcription factors were examined using western blotting (Fig. 4a). RANKL induced phosphorylation of p38MAPK, ERK and JNK, and triptolide (7 nM) inhibited their phosphorylation (Fig. 4a). Quantitative densitometry of phosphorylated p38MAPK, ERK, and JNK were shown in Fig. 4b. As shown, RANKL-induced phosphorylation of p38MAPK was markedly decreased at 15 minutes by triptolide. In addition, phosphorylated JNK and ERK were also decreased by triptolide. These results suggest that triptolide substantially inhibits RANKL-induced activation of MAP kinases, and thereby suppresses osteoclast differentiation and activation.

Inhibition of the RANK signaling pathway by triptolide. Mouse BMM cells were pretreated with triptolide (7 nM) for 30 minutes and stimulated with RANKL (50 ng/mL) for the indicated periods of time. a The phosphorylation levels (P) of JNK, p38MAPK, and ERK were analysed by Western blotting. The images are representative of three independent experiments. b Densitometry of blotting bands was analysed using a IMT i-Solution. The graphs are representative for the independent experiments
Triptolide suppresses Ti particle-induced osteolysis in a mouse calvarial model
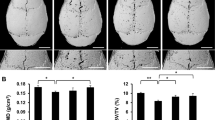
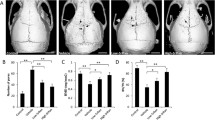
Having established that triptolide inhibits osteoclast differentiation and activity, we attempted to determine whether triptolide suppresses Ti particle-induced osteolysis in a mouse calvarial model. In the Ti PBS group, Ti particle induced osteolysis was apparent in CT images, whereas in Ti triptolide groups, Ti particle-induced osteolysis was substantially reduced by 16 and 32 μg/kg/day triptolide, respectively (Fig. 5a). Histology consistently showed that Ti injected bone resorption was dramatically reduced in a dose-dependent manner by treatment with triptolide (Fig. 5b). In TRAP staining, the Ti triptolide group (32 μg/kg/day) showed a dramatic reduction of TRAP positive osteoclasts, suggesting that triptolide also inhibits osteoclast formation in vivo. These results clearly demonstrate that triptolide suppresses Ti particle-induced osteolysis through suppression of osteoclast formation in mice.

μCT and histomorphometric analysis of calvaria. a Mouse calvaria were implanted with Ti particles, and five mice in each group were perorally administrated with 16 μg/kg/day (Ti+Tp16) or 32 μg/kg/day (Ti+Tp32) of triptolide. Ten days after feeding, calvaria were fixed and analysed with μCT. The images are representative of five calvaria from each group (n = 5). Bone density was measured from equal areas of calvaria. Lowest bone density was set to 1, and relative density is shown in the graph (*p < 0.02). b Calvaria prepared as in a were sectioned and stained with H&E (upper panel) and TRAP (lower panel). Average numbers of osteoclasts in slides (n = 3) were counted using a digitalized image analyser (IMT i-Solution) (right panel). *p < 0.05 and **p < 0.01
Discussion
In pathological conditions such as rheumatoid arthritis, enhanced activation and differentiation of osteoclasts induces bone loss and destruction. Triptolide has been shown to relieve or cure a wide variety of diseases [44]. In particular, triptolide inhibits collagen-induced arthritis through suppression of differentiation of Th17 cells [5]. In this study, we demonstrate that triptolide inhibits osteoclast differentiation and function through suppression of RANK signaling pathways and RANKL expression in osteoblasts. Since triptolide substantially suppressed osteoclast differentiation in BMM cell culture (Fig. 1a and b) as well as in a co-culture with osteoblasts and BMM cells (Fig. 2), it may target both osteoclasts and osteoblasts. In a recent study, Liu et al. demonstrated that triptolide prevents bone destruction in a collagen-induced arthritis model by targeting the RANKL/RANK/OPG signal pathway [43]. In human fibroblast-like synovial cells, triptolide inhibited expression of RANKL, but induced up-regulated expression of OPG. We consistently observed a trend showing suppression of RANKL and up-regulation of OPG expression in calvarial osteoblasts stimulated with vitamin D3 and PGE2 (Fig. 2c). Therefore, in both synovial cells and osteoblasts, triptolide may inhibit expression of RANKL, thus contributing to suppression of osteoclast formation.
Treatment with 14 nM triptolide induced a significant cytotoxic effect in BMM cells (Suppl. Fig. 1). The cytotoxicity of triptolide has already been recognized in different cell types [6, 9, 14]. However, treatment with low concentrations of triptolide (2.8 and 7 nM) specifically inhibited osteoclast differentiation without inducing cytotoxicity (Figs. 1 and 2). These results suggested that triptolide had a narrow window of efficacy in inhibiting osteoclast differentiation of BMM cells and bone resorption by osteoclasts.
RANK signaling in response to RANKL stimulation plays a central role in regulating differentiation and function of osteoclasts. Since triptolide significantly inhibited osteoclast differentiation of BMM cells in response to RANKL, it could be targeting RANK signaling pathway(s). As expected, triptolide substantially inhibited RANKL-induced activation of p38MAPK, ERK, and JNK (Fig. 4a). Furthermore, bone resorption by osteoclasts was also dramatically reduced by triptolide (Fig. 3), suggesting that triptolide may target RANK signaling pathways during various stages of osteoclast differentiation and activation. Substantial efforts have been made to define molecular targets of triptolide. A calcium channel polycystin-2 and a 90-kDa nuclear protein were identified as potential molecular targets of triptolide [45, 46]. More recently, Titov et al. reported that triptolide covalently binds to XPB/ERCC3, a subunit of the transcription factor TFIIH, and inhibits its DNA-dependent ATPase activity, which leads to inhibition of RNA polymerase II-mediated transcription and likely nucleotide excision repair [47]. Although we could not define a direct target molecule of triptolide in osteoclasts, our data suggest that triptolide may target RANKL-induced MAP kinase signaling pathways. A recent study reported that RANK expression was decreased by triptolide [43], suggesting that RANK expression by M-CSF, PMA, or vitamin D3 also could be a target for triptolide.
Although inflammatory cytokines such as IL-1β, IL-6 and TNF-α, which are secreted by macrophages/phagocytes engulfing wear particles, directly and/or indirectly stimulate osteoclast formation and bone resorption [18, 20, 21], RANKL also mediates wear particle-mediated osteolysis [22, 23]. Since triptolide dramatically inhibited osteoclast differentiation induced by RANKL, it may suppress inflammatory bone loss such as wear debris induced osteolysis. In fact, triptolide significantly suppressed Ti particle-induced calvarial osteolysis in mice (Fig. 5a). These results suggest a possibility that osteoclast formation is suppressed by triptolide in vivo. The numbers of osteoclasts induced by Ti particles was dramatically reduced by triptolide (Fig. 5b). Therefore, our data demonstrated that RANK signaling was critical for Ti particle-induced osteoclast formation, and that triptolide suppressed Ti particle-induced osteolysis. Triptolide might have an effect on other cells or tissues in vivo, leading to suppression of Ti-induced bone erosion. For example, triptolide inhibited IL-17 production by Th17 cells, splenocytes, and CD4 T cells [5, 40, 41]. Triptolide-mediated suppression of IL-17 production by Th17 cells significantly inhibited collagen-induced arthritis [5]. Moreover, immunohistochemical analysis showed that peri-implant tissues were positive for IL-17, suggesting a role of IL-17 in wear debris-induced aseptic loosening of prosthetic joints [48]. Therefore, triptolide might inhibit Ti particle-induced osteolysis through suppression of IL-17. It is also possible that Ti particle-induced production of other cytokines, such as TNF-α [49–51] and pro-inflammatory interleukins, could be inhibited by triptolide leading to suppression of Ti particle-induced osteolysis. Further studies may be necessary to confirm whether production of other inflammatory cytokines can be inhibited by triptolide, and thus resulting in the suppression of osteolysis.
Triptolide-induced toxicity in the hepatic, renal, digestive, reproductive, and hematological systems prevents it from being widely used in clinical practice. To overcome this problem and expand narrow efficacy windows, numerous triptolide derivatives have been synthesized [12, 16, 52, 53]. C-14-hydroxyl substitution improves the water solubility of triptolide, while retaining its anticancer activity in vivo and in vitro [12]. In addition, other approaches have been used to reduce triptolide toxicity. Since triptolide is absorbed rapidly into the blood circulation (from 5.0 to 19.5 min after administration) followed by a short elimination half-life (from approximately 20 minutes to one hour) [54], rapid fluctuations in triptolide plasma concentration could be a cause of toxicity. Nanostructured lipid carriers, which allowed sustained release of triptolide after oral administration, reduced toxicity in male rats, suggesting that controlled release of triptolide can substantially reduce multiple tissue toxicity [54]. Therefore, triptolide could be a candidate drug for the treatment of osteolytic diseases. Whether non-toxic triptolide derivatives or sustained release of triptolide would significantly improve its efficacy in wear debris-induced osteolysis remains to be investigated.
References
Wang J, Wang A, Zeng H, Liu L, Jiang W, Zhu Y, Xu Y (2012) Effect of triptolide on T-cell receptor beta variable gene mRNA expression in rats with collagen-induced arthritis. Anat Rec 295(6):922–927
Xue M, Jiang ZZ, Liu JP, Zhang LY, Wang T, Wang H, Liu L, Zhou ZX (2010) Comparative study on the anti-inflammatory and immune suppressive effect of Wilforlide A. Fitoterapia 81(8):1109–1112
Kupchan SM, Court WA, Dailey RG Jr, Gilmore CJ, Bryan RF (1972) Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J Am Chem Soc 94(20):7194–7195
Wong KF, Yuan Y, Luk JM (2012) Tripterygium wilfordii bioactive compounds as anticancer and anti-inflammatory agents. Clin Exp Pharmacol Physiol 39(3):311–320
Wu Y, Cui J, Bao X, Chan S, Young DO, Liu D, Shen P (2006) Triptolide attenuates oxidative stress, NF-kappaB activation and multiple cytokine gene expression in murine peritoneal macrophage. Int J Mol Med 17(1):141–150
Qiu D, Kao PN (2003) Immunosuppressive and anti-inflammatory mechanisms of triptolide, the principal active diterpenoid from the Chinese medicinal herb Tripterygium wilfordii Hook. f. Drugs R D 4(1):1–18
Liu Q (2011) Triptolide and its expanding multiple pharmacological functions. Int Immunopharmacol 11(3):377–383
Xue X, Gong L, Qi X, Wu Y, Xing G, Yao J, Luan Y, Xiao Y, Li Y, Wu X, Chen M, Gu J, Ren J (2011) Knockout of hepatic P450 reductase aggravates triptolide-induced toxicity. Toxicol Lett 205(1):47–54
Li J, Jin J, Li M, Guan C, Wang W, Zhu S, Qiu Y, Huang M, Huang Z (2012) Role of Nrf2 in protection against triptolide-induced toxicity in rat kidney cells. Toxicol Lett 213(2):194–202
Ye X, Li W, Yan Y, Mao C, Cai R, Xu H, Yang X (2010) Effects of cytochrome P4503A inducer dexamethasone on the metabolism and toxicity of triptolide in rat. Toxicol Lett 192(2):212–220
Chen H, Chang X, Weng T, Zhao X, Gao Z, Yang Y, Xu H, Yang X (2004) A study of microemulsion systems for transdermal delivery of triptolide. J Control Release 98(3):427–436
Xu F, Shi X, Li S, Cui J, Lu Z, Jin Y, Lin Y, Pang J, Pan J (2010) Design, synthesis, and biological evaluation of novel water-soluble triptolide derivatives: Antineoplastic activity against imatinib-resistant CML cells bearing T315I mutant Bcr-Abl. Bioorg Med Chem 18(5):1806–1815
Fidler JM, Li K, Chung C, Wei K, Ross JA, Gao M, Rosen GD (2003) PG 490–88, a derivative of triptolide, causes tumor regression and sensitizes tumors to chemotherapy. Mol Cancer Ther 2(9):855–862
Aoyagi Y, Hitotsuyanagi Y, Hasuda T, Matsuyama S, Fukaya H, Takeya K, Aiyama R, Matsuzaki T, Hashimoto S (2008) Fluorination of triptolide and its analogues and their cytotoxicity. Bioorg Med Chem Lett 18(7):2459–2463
Zhou ZL, Yang YX, Ding J, Li YC, Miao ZH (2012) Triptolide: structural modifications, structure-activity relationships, bioactivities, clinical development and mechanisms. Nat Prod Rep 29(4):457–475
Chugh R, Sangwan V, Patil SP, Dudeja V, Dawra RK, Banerjee S, Schumacher RJ, Blazar BR, Georg GI, Vickers SM, Saluja AK (2012) A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci Transl Med 4(156):156ra139
Dumbleton JH, Manley MT, Edidin AA (2002) A literature review of the association between wear rate and osteolysis in total hip arthroplasty. J Arthroplasty 17(5):649–661
Goldring SR, Schiller AL, Roelke M, Rourke CM, O’Neil DA, Harris WH (1983) The synovial-like membrane at the bone-cement interface in loose total hip replacements and its proposed role in bone lysis. J Bone Joint Surg Am 65(5):575–584
Jin S, Park JY, Hong JM, Kim TH, Shin HI, Park EK, Kim SY (2011) Inhibitory effect of (-)-epigallocatechin gallate on titanium particle-induced TNF-alpha release and in vivo osteolysis. Exp Mol Med 43(7):411–418
Merkel KD, Erdmann JM, McHugh KP, Abu-Amer Y, Ross FP, Teitelbaum SL (1999) Tumor necrosis factor-alpha mediates orthopedic implant osteolysis. Am J Pathol 154(1):203–210
Shanbhag AS, Jacobs JJ, Black J, Galante JO, Glant TT (1995) Cellular mediators secreted by interfacial membranes obtained at revision total hip arthroplasty. J Arthroplasty 10(4):498–506
Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TP (2007) The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res 454:251–261
Ren W, Wu B, Peng X, Hua J, Hao HN, Wooley PH (2006) Implant wear induces inflammation, but not osteoclastic bone resorption, in RANK(−/−) mice. J Orthop Res 24(8):1575–1586
Ingham E, Fisher J (2005) The role of macrophages in osteolysis of total joint replacement. Biomaterials 26(11):1271–1286
Kwak HB, Lee BK, Oh J, Yeon JT, Choi SW, Cho HJ, Lee MS, Kim JJ, Bae JM, Kim SH, Kim HS (2010) Inhibition of osteoclast differentiation and bone resorption by rotenone, through down-regulation of RANKL-induced c-Fos and NFATc1 expression. Bone 46(3):724–731
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423(6937):337–342
Cappellen D, Luong-Nguyen NH, Bongiovanni S, Grenet O, Wanke C, Susa M (2002) Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NFkappa B. J Biol Chem 277(24):21971–21982
Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J (2001) Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 20(6):1271–1280
Hotokezaka H, Sakai E, Kanaoka K, Saito K, Matsuo K, Kitaura H, Yoshida N, Nakayama K (2002) U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast-like cells. J Biol Chem 277(49):47366–47372
Matsuo K, Galson DL, Zhao C, Peng L, Laplace C, Wang KZ, Bachler MA, Amano H, Aburatani H, Ishikawa H, Wagner EF (2004) Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem 279(25):26475–26480
Nakashima T, Takayanagi H (2009) Osteoimmunology: crosstalk between the immune and bone systems. J Clin Immunol 29(5):555–567
Zhao Q, Wang X, Liu Y, He A, Jia R (2010) NFATc1: functions in osteoclasts. Int J Biochem Cell Biol 42(5):576–579
Park JY, Bae MA, Cheon HG, Kim SS, Hong JM, Kim TH, Choi JY, Kim SH, Lim J, Choi CH, Shin HI, Kim SY, Park EK (2009) A novel PPARgamma agonist, KR62776, suppresses RANKL-induced osteoclast differentiation and activity by inhibiting MAP kinase pathways. Biochem Biophys Res Commun 378(3):645–649
Park EK, Lee YE, Choi JY, Oh SH, Shin HI, Kim KH, Kim SY, Kim S (2004) Cellular biocompatibility and stimulatory effects of calcium metaphosphate on osteoblastic differentiation of human bone marrow-derived stromal cells. Biomaterials 25(17):3403–3411
Faccio R, Takeshita S, Zallone A, Ross FP, Teitelbaum SL (2003) c-Fms and the alphavbeta3 integrin collaborate during osteoclast differentiation. J Clin Invest 111(5):749–758
Battaglino R, Kim D, Fu J, Vaage B, Fu XY, Stashenko P (2002) c-myc is required for osteoclast differentiation. J Bone Miner Res 17(5):763–773
Vukosavic S, Stefanis L, Jackson-Lewis V, Guegan C, Romero N, Chen C, Dubois-Dauphin M, Przedborski S (2000) Delaying caspase activation by Bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 20(24):9119–9125
Choi YA, Lim J, Kim KM, Acharya B, Cho JY, Bae YC, Shin HI, Kim SY, Park EK (2010) Secretome analysis of human BMSCs and identification of SMOC1 as an important ECM protein in osteoblast differentiation. J Proteome Res 9(6):2946–2956
Wu Y, Wang Y, Zhong C, Li Y, Li X, Sun B (2003) The suppressive effect of triptolide on experimental autoimmune uveoretinitis by down-regulating Th1-type response. Int Immunopharmacol 3(10–11):1457–1465
Wang Y, Jia L, Wu CY (2008) Triptolide inhibits the differentiation of Th17 cells and suppresses collagen-induced arthritis. Scand J Immunol 68(4):383–390
Li Y, Yu C, Zhu WM, Xie Y, Qi X, Li N, Li JS (2010) Triptolide ameliorates IL-10-deficient mice colitis by mechanisms involving suppression of IL-6/STAT3 signaling pathway and down-regulation of IL-17. Mol Immunol 47(15):2467–2474
Chen ZH, Qin WS, Zeng CH, Zheng CX, Hong YM, Lu YZ, Li LS, Liu ZH (2010) Triptolide reduces proteinuria in experimental membranous nephropathy and protects against C5b-9-induced podocyte injury in vitro. Kidney Int 77(11):974–988
Liu C, Zhang Y, Kong X, Zhu L, Pang J, Xu Y, Chen W, Zhan H, Lu A, Lin N (2013) Triptolide prevents bone destruction in the collagen-induced arthritis model of rheumatoid arthritis by targeting RANKL/RANK/OPG signal pathway. Evid Based Complement Alternat Med 2013:626038
Han R, Rostami-Yazdi M, Gerdes S, Mrowietz U (2012) Triptolide in the treatment of psoriasis and other immune-mediated inflammatory diseases. Br J Clin Pharmacol 74(3):424–436
Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, Crews CM (2007) Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci USA 104(11):4389–4394
McCallum C, Kwon S, Leavitt P, Shen DM, Liu W, Gurnett A (2007) Triptolide binds covalently to a 90 kDa nuclear protein. Role of epoxides in binding and activity. Immunobiology 212(7):549–556
Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO (2011) XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol 7(3):182–188
Hercus B, Saeed S, Revell PA (2002) Expression profile of T cell associated molecules in the interfacial tissue of aseptically loosened prosthetic joints. J Mater Sci Mater Med 13(12):1153–1156
Al-Saffar N, Khwaja HA, Kadoya Y, Revell PA (1996) Assessment of the role of GM-CSF in the cellular transformation and the development of erosive lesions around orthopaedic implants. Am J Clin Pathol 105(5):628–639
Cano E, Mahadevan LC (1995) Parallel signal processing among mammalian MAPKs. Trends Biochem Sci 20(3):117–122
Crotti TN, Smith MD, Findlay DM, Zreiqat H, Ahern MJ, Weedon H, Hatzinikolous G, Capone M, Holding C, Haynes DR (2004) Factors regulating osteoclast formation in human tissues adjacent to peri-implant bone loss: expression of receptor activator NFkappaB, RANK ligand and osteoprotegerin. Biomaterials 25(4):565–573
Zhou B, Li X, Tang H, Miao Z, Feng H, Li Y (2011) Total synthesis of novel D-ring-modified triptolide analogues: structure-cytotoxic activity relationship studies on the D-ring of triptolide. Org Biomol Chem 9(9):3176–3179
Kupchan SM, Schubert RM (1974) Selective alkylation: a biomimetic reaction of the antileukemic triptolides? Science 185(4153):791–793
Zhang C, Peng F, Liu W, Wan J, Wan C, Xu H, Lam CW, Yang X (2014) Nanostructured lipid carriers as a novel oral delivery system for triptolide: induced changes in pharmacokinetics profile associated with reduced toxicity in male rats. Int J Nanomedicine 9:1049–1063
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MISP) (2008-0062282), and Kyungpook National University Research Fund, 2013.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ju Ang Kim and Hye Jung Ihn contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 6
Cell viability of mouse BMM cells with triptolide. The effect of triptolide on BMM cells viability was determined using the MTT cytotoxicity assay. BMM cells were cultured in α-MEM containing 30 ng/mL M-CSF with or without 2.8, 7, or 14 nM of triptolide. (GIF 19 kb)
Rights and permissions
About this article

Cite this article
Kim, J.A., Ihn, H.J., Park, JY. et al. Inhibitory effects of triptolide on titanium particle-induced osteolysis and receptor activator of nuclear factor-κB ligand-mediated osteoclast differentiation. International Orthopaedics (SICOT) 39, 173–182 (2015). https://doi.org/10.1007/s00264-014-2596-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00264-014-2596-3