
Abstract
Immune checkpoint inhibitors (ICIs) are used in cancer immunotherapy to block programmed death-1 and cytotoxic T-lymphocyte antigen 4, but the response rate for ICIs is still low and tumor cell heterogeneity is considered to be responsible for resistance to immunotherapy. Tumor-infiltrating lymphocytes (TILs) have an essential role in the anti-tumor effect of cancer immunotherapy; however, the specificity of TILs in renal cell carcinoma (RCC) is elusive. In this study, we analyzed a 58-year-old case with clear cell RCC (ccRCC) with the tumor showing macroscopic and microscopic heterogeneity. The tumor was composed of low-grade and high-grade ccRCC. A tumor cell line (1226 RCC cells) and TILs were isolated from the high-grade ccRCC lesion, and a TIL clone recognized a novel neoantigen peptide (YVVPGSPCL) encoded by a missense mutation of the tensin 1 (TNS1) gene in a human leukocyte antigen-C*03:03-restricted fashion. The TNS1 gene mutation was not detected in the low-grade ccRCC lesion and the TIL clone did not recognized low-grade ccRCC cells. The missense mutation of TNS1 encoding the S1309Y mutation was found to be related to cell migration by gene over-expression. These findings suggest that macroscopically and microscopically heterogenous tumors might show heterogenous gene mutations and reactivity to TILs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There were an estimated 73,750 new cases of kidney and renal pelvis cancer and 14,830 deaths due to the disease in the USA in 2020 [1]. Although the 5-year survival rate of patients with localized kidney cancer is more than 90%, approximately 30% of patients are diagnosed with metastatic disease [1,2,3]. For patients with metastatic renal cell carcinoma (RCC), molecular targeting therapy with vascular endothelial growth factor- and mammalian target of rapamycin-targeting agents has been used for more than 10 years [3,4,5,6,7,8]. Recently, treatment of metastatic RCC has changed dramatically, as with other carcinomas, following the development of immune checkpoint inhibitors [9,10,11,12]. Although an anti-programmed death-1 (PD-1) antibody was approved for patients with metastatic RCC, a PD-1 inhibitor was also shown to be effective for these patients, even in those with less than 1% programmed death ligand 1 (PD-L1) expression [9]. Originally, RCC was identified as a representative tumor for which immune therapy with interleukin (IL)-2 and interferon (IFN)-α was beneficial, similar to malignant melanoma [13,14,15]. However, it is still unknown why RCC is an immune-sensitive tumor [16,17,18]. Recent studies suggested that RCC might be immunogenic because of high levels of neoantigens encoded by indel mutations and high rates of endogenous retroviruses [19, 20]. However, the roles of tumor-infiltrating lymphocytes (TILs) in RCC are still elusive.
Cytotoxic T lymphocytes (CTLs) are powerful effectors of the immune system, providing protection against pathogens and cancers. CTLs recognize several types of tumor-associated antigens (TAAs), which are neoantigens encoded by gene mutations, viral antigens, over-expressed antigens, and cancer-testis antigens [21]. We previously reported an antigenic peptide encoded by the over-expressed antigen HIFPH3, and HIFPH3 peptide-specific CTLs could be induced from the peripheral blood of RCC patients [22]. Other studies have revealed that RCC-reactive CTLs can be isolated from peripheral blood [23,24,25,26]; however, there is a limited number of TAAs recognized by the TILs of RCC. Tumor-infiltrating CTLs, referred to as CD8+ TILs, have been suggested to have a prognostic role for immunotherapy in some solid tumors [27,28,29]. However, their role in RCC is controversial, unlike in other tumors [30]. Some studies have revealed that a high density of CD8+ TILs is associated with a poor clinical outcome in RCC [31,32,33]. Bystander TILs that do not recognize cancer cells might be one of the reasons for this phenomenon [34]. To clarify the specificity of TILs in RCC, we investigated whether TILs isolated from RCC recognize a tumor-specific antigen, and identified a novel neoantigen encoded by a missense mutation in the tensin 1 (TNS1) gene that has a role in the migration of RCC cells.
Materials and methods
Ethics statement
In this study, we used material derived from a single patient. The experimental protocol was approved by the institutional-review board of Sapporo Medical University (approval no. 282–152), and written informed consent was obtained from the patient.
Patient and establishment of an RCC cell line
Primary RCC cells and peripheral blood were obtained from a 58-year-old female patient with RCC cT4N2M1 (human leukocyte antigen [HLA]-A*02:01, A*24:02, B*35:01, B*40:01, C*03:03, C*15:02, DRB1*04:05, DRB1*15:01) who underwent left nephrectomy at Sapporo Medical University Hospital. The tumor was heterogenous, and there was a reddish lesion (red tumor) and a yellowish lesion (yellow tumor). Tissues from the red and yellow tumors were obtained, minced with sterile scissors, and incubated in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA) containing 1 mg/mL Liberase™ (Roche, Basel, Switzerland) at 37 °C for 20 min. Single cell suspensions were collected by filtering through a 70-μm nylon cell strainer (Corning, Inc., Corning, NY, USA). The cells were washed 3 times with sterile phosphate-buffered saline and recovered by centrifugation. Some single cell suspensions were stored at − 80 °C for future analyses. A single cell suspension was cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (Biosera, Kansas City, MO, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. A human RCC cell line, 1226 RCC, was established from only the red tumor. The 1226 RCC cells could grow stably for more than 50 passages in vitro. An Epstein-Barr virus (EBV) lymphoblastoid cell line (1226 EBV-B cells) was established by using the culture supernatant of the B95.8 cell line.
Establishment of autologous TIL clones
Autologous TIL clones were generated from single cell suspensions from the primary tumor. A single cell suspension (5.0 × 105 cells/well) was placed in each well of a 96-well round-bottom microplate with complete medium plus 5,000 IU/mL of recombinant human (rh) IL-2 (PeproTech, Cranbury, NJ, USA) and the cultures were maintained at 37 °C in 5% CO2. Complete medium consisted of AIM-V medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10 mM HEPES (Thermo Fisher Scientific), 2 mM glutamine (Glutamax™-1; Thermo Fisher Scientific), 5.5 × 10–5 mol/L β-mercaptoethanol, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% human serum. On days 8, 12, and 19, the cultured cells were stimulated by adding 5.0 × 104 to 5.0 × 105 irradiated (100 Gy) autologous tumor cells (1226 RCC). From day 8, the TILs were fed using complete medium containing 100 IU/mL rhIL-2. On day 22, the TILs were analyzed by flow cytometry, and CD137+ cells were single cell sorted using a FACS Aria II (BD Biosciences, San Jose, CA, USA). Establishment of TIL clone cells was performed as described previously [35].
IFNγ enzyme-linked immunospot (ELISPOT) assay
IFNγ production by the TIL clones was assessed using an ELISPOT assay (Human IFNγ ELISPOT Set; BD Biosciences) as described previously [36]. TILs or TIL clone cells were used at 1.0–2.0 × 104 cells/mL per well. Target cells at 5.0 × 104 were added to the corresponding wells. HEK293T-HLA*C03:03 cells were used to transiently express one of a series of tandem minigene (TMG) constructs. HEK293T-HLA*C03:03 cells were preincubated at room temperature for 2 h with 20 μmol/L to 200 pmol/L of wild-type or mutated TNS1 peptide or irrelevant EBV peptide. For blocking HLA, the target cells were preincubated with an anti-pan HLA class 1 monoclonal antibody (mAb) (W6/32), anti-HLA-A2 mAb (BB7.2), anti-HLA-A24 mAb (C7709A2.6), anti-HLA-BC mAb (B1.23.2), or anti-HLA-DR mAb (L243) for 30 min.
Plasmid construction, transfection, and western blot analysis
Human HLA-B*35:01:01, HLA-B*40:01:02, HLA-C*03:03:01, and HLA-C*15:02:01 cDNAs in pcDNA3.1 plasmids were obtained from RIKEN BRC (Tsukuba, Japan) through the National BioResource Project of MEXT/AMED, Japan [37]. HLA genes were subcloned into a retroviral pMXs-Puro vector (a kind gift from Dr. T. Kitamura, Tokyo, Japan) to obtain stable transfectants. HLA-B*35:01:01, HLA-C*03:03:01, and HLA-C*15:02:01 were subcloned into the BglII and NotI sites, and HLA-B*40:01:02 was subcloned into the HindIII and NotI sites.
TNS1 cDNA was cloned from cDNA isolated from 1226 RCC cells by reverse transcription polymerase chain reaction (RT-PCR). Isolation of total RNA and preparation of cDNA were performed as described previously [38]. The TNS1 gene was amplified as 4 fragments using PrimeSTAR® GXL according to the manufacturer’s instruction (TakaraBio, Kusatsu, Japan). Primer pairs for the TNS1 gene were 5′-TAGCTAGTTAATTAAGCCACCATGAGTGTGAGCCGGACCAT-3′ and 5′-ATGGCACAGGTGTGGAAC-3′ for fragment 1, 5′-GTTCCACACCTGTGCCAT-3′ and 5′-TATAGCCAGATCTGGACTGG-3′ for fragment 2, 5′-CCAGTCCAGATCTGGCTATA-3′ and 5′-GTGTCCTGGACAAACTTCAC-3′ for fragment 3, and 5′-GTGAAGTTTGTCCAGGACAC-3′ and 5′-TGTGCTGGCGGCCGCTCATCTCTTTTGGCCGGCAT-3′ for fragment 4. Fragments 1 and 4 included a PacI site and a NotI site (underlined), respectively. The 4 fragments were subcloned into the pMXs-Puro vector using an In-Fusion HD Cloning Kit (Takara Bio). The sequence of the TNS1 gene was confirmed by direct sequencing. The TNS1 gene derived from the 1226 RCC cells was the 1309Y type. To establish a 1309S TNS1 gene, we performed mutagenesis using PCR. The 1309S TNS1 gene was amplified as 2 fragments using the primer pairs 5′-TAGCTAGTTAATTAAGCCACCATGAGTGTGAGCCGGACCAT-3′ and 5′-GGAACCACAGATCCAGACC-3′, and 5′-GGTCTGGATATGTGGTTCC-3′ and 5′-TGTGTGGCGGCCGCTCATCTCTTTTGGCCGGCAT-3′ (underlined text indicates the 1309 Ser residue). Both fragments were subcloned into the pMXs-Puro vector and confirmed by direct sequencing.
Transduction of genes into cells was carried out by a retrovirus-mediated method as described previously using PLAT-A amphotropic packaging cells (a kind gift from Dr. T. Kitamura, Tokyo, Japan) [39]. To establish stable transformants, the infected cells were selected with 1 μg/mL puromycin (Takara Bio). To confirm the expression of TNS1 protein, western blot analysis was performed as described previously [40]. A rabbit anti-TNS1 mAb (Thermo Fisher Scientific) was used at 500-times dilution.
Whole exome sequencing
For library construction, genomic DNA was extracted from 1226 RCC cells and peripheral blood mononuclear cells (PBMCs) of the patient. Library construction, exome capture of coding genes, and next-generation sequencing were performed at Macrogen Japan Corp. (Kyoto, Japan) as follows. Enrichment of exome sequences was performed for each library using a SureSelect V6-post Kit following the manufacturer’s instructions (Agilent Technologies, Tokyo, Japan). Paired-end sequencing, resulting in 151-bp reads from both ends of each fragment, was performed for the whole exomes using a NovaSeq 6000 (Agilent Technologies). More than 18 billion bases of sequence data were obtained for each sample. Sequence data were mapped to the reference human genome sequence, including 93,457 single nucleotide polymorphisms (SNPs) and 14,663 indels in 1226 RCC cells and 99,024 SNPs and 15,669 indels in PBMCs. More than 60 million bases of target DNA were analyzed in 1226 RCC cells and PBMCs, and an average of 147 to 148 reads were obtained at each base in the tumor and PBMC samples.
Reads were aligned to the NCBI human reference genome (hg19) using BWA [41]. Picard was applied to mark duplicates and we used the Genome Analysis Toolkit (GATK) Indel Realigner to improve alignment accuracy. Somatic single-nucleotide variants from 101 lung squamous cell carcinoma samples with matched noncancer control samples were called using MuTect [42]. GATK Haplotype Caller was also used for indel detection. All variants were annotated with information from several databases using ANNOVAR [43].
Construction of a TMG library
Nonsynonymous mutations from 1226 RCC cells were identified from the whole exome sequencing data. TMG constructs that encoded polypeptides containing 10–11 identified mutated amino acid residues that were flanked on their N- and C- termini by 12 amino acids and with a Kozak consensus sequence at the start and the termination codon TGA at the end were synthesized (Integrated DNA Technologies, Coralville, IA, USA) (summarized in Table S1). The restriction sites NheI, HindIII, BamHI, EcoRI, NotI, and XhoI were included in the TMGs. TMGs were subcloned into pcDNA3.1(+) (Thermo Fisher Scientific) at the NheI and XhoI sites and confirmed by direct sequencing.
Analysis of the CDR3 sequence of TIL clone Q1 and T cell receptor (TCR) repertoire analysis
To clone TCRα/β genes, 5′-rapid amplification of cDNA ends (RACE) was performed using a SMARTer RACE cDNA Amplification Kit (Takara Bio). For the first round of PCR, cDNA was amplified using the supplied 5′-RACE primer and a 3′-TCRα untranslated region primer (5′-CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAGAGT-3′). The second round of PCR was performed using a modified 5′-RACE primer (5′-GTGTGGTGGTACGGGAATTCAAGCAGTGGTATCAACGCAGAGT-3′) and a 3′-TCRα primer (5′-ACCACTGTGCTGGCGGCCGCTCAGCTGGACCACAGCCGCAGCG-3′). TCR gene structure was analyzed by direct sequencing.
Genomic DNAs were isolated from primary tumor single cell suspensions and PBMCs. Then, TCR repertoire was analyzed by Repertoire Genesis (Osaka, Japan).
DNA sequencing of the TNS1 gene
Extraction of total nucleic acids from formalin-fixed paraffin-embedded tissues of the red and yellow tumors was performed using a RecoverAll™ Total Nucleic Acid Isolation Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. RT-PCR was performed using the following primer pair: 5′-CCAGTCTCCAGAAGCCACATC-3′ and 5′-GGTAGTTTCTCTGGGCAGACG-3′ for TNS1 (expected PCR product, 207 bp). DNA sequencing was performed by direct sequencing.
Antibodies
The primary antibodies used were anti-HLA class 1 mAb (W6/32; isotype IgG2a), anti-HLA-A24 mAb (C7709A2.6; IgG2a), anti-HLA-A2 mAb (BB7.2; IgG2b), anti-HLA-BC mAb (B1.23.2; IgG2b), and anti HLA-DR mAb (L243; IgG2a); these hybridomas were kindly donated or purchased from the American Type Culture Collection (Manassas, VA, USA). Anti-HLA-B mAb (JOAN-1, IgG1; Thermo Fisher Scientific), anti-HLA-C mAb (DT9, IgG2b; BD Biosciences), anti-CD8 mAb (APC, IgG1; MBL, Nagoya, Japan), anti-CD4 mAb (FITC; BD Biosciences), anti-CD137 mAb (VioBright FITC, Miltenyi Biotec, Tokyo, Japan), and rabbit anti-PD-L1 mAb (clone E1L3N; Cell Signaling Technology, Danvers, MA, USA) were also used. Anti-mouse IgG + IgM (FITC; KPL) was used as a secondary antibody.
Statistical analysis
Student’s t-test was used to compare two groups. P < 0.05 was considered to indicate statistical significance.
Results
Macroscopic, microscopic, and immunological heterogeneity of an RCC case
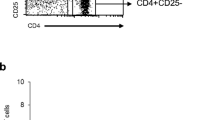
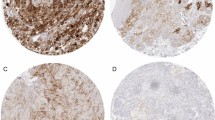
In this study, we analyzed a 58-year-old female patient with stage 4 RCC. Macroscopic findings revealed that the tumor was heterogeneous and consisted of a yellowish lesion and a reddish lesion (yellow tumor and red tumor, respectively) (Fig. 1A). Pathohistological findings revealed that the yellow tumor was clear cell RCC with Fuhrman nuclear grade 1. The red tumor was clear cell RCC with Fuhrman nuclear grade 2. Immunohistochemical staining revealed that the red and yellow tumors were positive for HLA class 1. Interestingly, the red tumor showed a relatively higher number of infiltrating CD8+ T cells compared with the yellow tumor (Fig. 1A). The red tumor showed the infiltration of PD-1+ cells, but none were detected in the yellow tumor. RCC cells in the red tumor expressed PD-L1 weakly. On the other hand, RCC cells in the yellow tumor did not express PD-L1 (Figure S2). PD-L1 is also expressed in stromal macrophage in the red tumor suggesting that TILs in the red tumor secrete interferon-γ(IFNγ). TILs from the yellow and red tumors were analyzed by flow cytometry. TILs were detected at 3.7% of primary RCC cells from the red tumor; however, TILs could not be detected from the yellow tumor (Fig. 1b). TILs from the red tumor included CD8+ T cells and CD4+ T cells (Fig. 1b and S2).

Macroscopic, pathological, and immunological findings of a clear cell RCC case. a Macroscopic and microscopic images of the tumor. Macroscopically, the tumor showed heterogeneity with a reddish lesion (red tumor) and yellowish lesion (yellow tumor). An RCC cell line, 1226 RCC cells, was established from the red tumor. The yellow tumor showed clear cell RCC with Fuhrman nuclear grade 1, and the red tumor showed Fuhrman nuclear grade 2. CD8+ T cell infiltration and HLA class 1 expression were examined by immunohistochemical staining. Dotted lines indicate the epithelial-subepithelial margin. HE: hematoxylin and eosin staining. Original magnification × 250 (upper image) and × 500 (lower image); bar = 200 μm. b TIL analysis by flow cytometry. Flow cytometry analysis of TILs in the red and yellow tumors. TILs from the red tumor contained 27.8% CD4+ T cells and 44.1% CD8+ T cells in 3.7% of putative lymphocytes gated by forward scatter and side scatter using tumor single cell suspensions. No TILs were detected from the yellow tumor
Isolation of an HLA-C*03:03-restricted TIL clone
To facilitate immunological analysis, we established a novel RCC cell line (1226 RCC) from the red tumor. The 1226 RCC cell line grew stably for more than 50 passages. Bulk TILs recognized the 1226 RCC cells by an IFNγ ELISPOT assay compared with negative control K562 cells (Figure S1). To analyze further the TILs at the clonal level, we isolated CD137+ (4-1BBL) cells. TILs were stimulated by co-culture with 1226 RCC cells and then analyzed. The CD137+ rate of TILs with stimulation by 1226 RCC cells was 0.4%, whereas it was 0.1% without stimulation. We then isolated CD137+ cells from TILs stimulated by 1226 RCC cells and established 29 TIL clones (Fig. 2a); 22 of which recognized 1226 RCC cells by an IFNγ ELISPOT assay. The TIL clone Q1 showed reactivity to 1226 RCC cells that was inhibited by an anti-HLA class 1 mAb and anti-HLA-BC mAb, indicating that the Q1 clone recognized 1226 RCC cells in an HLA-B- or HLA-C-restricted manner (Fig. 2b). The HLA-B and -C genotypes of the patient were HLA-B*35:01/B*40:01 and -C*03:03/C*15:02, and no allele-specific antibody was available. Thus, we overexpressed HLA-B or -C genes in 1226 RCC cells and examined the reactivity of the Q1 clone (Fig. 2c). Over-expression of HLA-C*03:03 increased the number of IFNγ spots, whereas over-expression of other HLA-B or -C genes decreased the number of IFNγ spots. The IFNγ spots were inhibited by an anti-HLA-BC antibody, indicating that the Q1 TIL clone was HLA-C*03:03-restricted (Fig. 2c).

Establishment of the reactive 1226 RCC TIL clone. a. TIL clone establishment method. TILs obtained from the red tumor were stained with anti-CD8 and anti-CD137 antibodies. TILs were co-cultured with 1226 RCC cells before FACS analysis. TILs without co-culture were used as a negative control. CD137+ cells were single cell-sorted using a cell sorter. After several weeks of in vitro culture, 116 of 384 clones showed cell growth. b IFNγ ELISPOT assay of the Q1 TIL clone. Q1 TIL clones were co-cultured with 1226 RCC cells and analyzed by an IFNγ ELISPOT assay. Anti-HLA-pan class 1 (W6/32), anti-HLA-A24 (C7709A2.6), anti-HLA-A2 (BB7.2), anti-HLA-BC (B1.23.2), and anti-HLA-DR (L243) antibodies were used to assess the specific reactivity of the Q1 TIL clone. 1226 EBV-B cells and K562 cells were used as negative controls. IFNγ spots are shown as mean ± standard error of the mean (SEM; n = 3), and p values were calculated using a paired t test; *p < 0.01. c IFNγ ELISPOT assay of the Q1 TIL clone using HLA allele-specific over-expressing 1226 RCC cells. HLA-B*35:01, B*40:01, C*03:03, and C*15:02 were stably over-expressed in 1226 RCC cells. Q1 TIL clone reactivity for HLA alleles over-expressed in 1226 RCC cells was examined by an IFNγ ELISPOT assay. An anti-HLA-BC (B1.23.2) antibody was used to confirm specificity. IFNγ spots are shown as the mean ± SEM (n = 3), and p values were calculated using a paired t test; *p < 0.01
RCC-reactive TIL clones recognize a neoantigen derived from a mutated TNS1 gene
We hypothesized that the Q1 TIL clone recognized a neoantigen derived from a cancer-specific gene mutation; therefore, we performed whole exome sequencing using 1226 RCC cells. Whole exome sequencing analysis revealed that 1226 RCC cells harbored 340 SNPs and 149 indel mutations compared with the patient’s PBMCs as a control, and these mutations encoded 87 missense mutations and 8 frameshifts (Fig. 3a). To address the immunogenicity of all gene mutations, we synthesized 9 TMGs (TMG-1–9) that covered all missense mutations and frameshifts (Fig. 3a and Table S1). Each TMG encoded a series of nonsynonymous mutations with 36 base pairs (12 amino acids) and 5′- and 3′-flanking sequences. A representative TMG (TMG-2) is shown in Fig. 3b. HEK293T cells stably over-expressing HLA-C*03:03 (293T-C*03:03 cells) were transiently transfected with the 9 TMG constructs, and the reactivity of the Q1 clone was analyzed by an IFNγ ELISPOT assay. The Q1 clone recognized 293T-C*03:03 cells transfected with TMG-2 (Fig. 3c), which encoded 11 minigenes (G12–G22). We further constructed additional TMGs by exchanging minigenes between TMG-2 and TMG-1 (Fig. 3d), and the reactivity of the Q1 TIL clone was examined (Fig. 3e). Finally, the Q1 TIL clone showed positive reactivity for gene #16 (TNS1) (Fig. 3f, g), which harbored a TNS1 missense mutation (a single C to A transversion at nucleotide 3926 of the TNS1 coding region), resulting in a substitution of serine to tyrosine at position 1309 of wild-type TNS1 protein (Fig. 3g).

Identification of the tumor antigen recognized by the Q1 TIL clone. a Schematic summary of whole exome sequencing of 1226 RCC cells and design of TMGs. A total of 340 SNPs and 149 indel mutations were detected in the genome of 1226 RCC cells compared with the genome from the patient’s PBMCs. The mutations encoded 87 missense mutations and 8 frameshift mutations. The mutations were designed into 9 TMGs. b Design of TMGs. The gene started with the Kozak consensus sequence followed by a start codon. For missense mutations, TMGs encoded polypeptides containing mutated amino acid residues flanked on their N- and C-termini with 12 amino acids across restriction enzyme sites. TMGs were cloned into the pcDNA3.1 plasmid. For frameshift mutations, the TMGs encoded polypeptides of frameshifted amino acid residues flanked on their N-terminus with 12 amino acids. c The Q1 TIL clone recognizes TMG-2. Each TMG was transiently transfected into 293 T-C*03:03 cells and the reactivity of the Q1 TIL clone was assessed by an IFNγ ELISPOT assay. 1226 RCC cells were used as a positive control. IFNγ spots are shown as mean ± SEM (n = 3). d, e, f, g. The Q1 TIL clone recognizes gene 16 in an HLA-C*03:03-restricted manner. To narrow down the candidate gene, several TMGs were constructed (D and F). The reactivity of the Q1 TIL clone was assessed by an IFNγ ELISPOT assay (E and G). 1226 RCC cells were used as a positive control. IFNγ spots are shown as mean ± SEM (n = 3), and p values were calculated using a paired t test; *p < 0.01
To identify the epitope, we synthesized peptides of different lengths including the missense mutation. 293T-C*03:03 cells were pulsed with candidate peptides and the reactivity of the Q1 TIL clone was examined (Fig. 4a). The Q1 TIL clone recognized the 12Y, 11Y, 10Y, and 9Y peptides, but not the 8VL or 9GC peptides. Thus, we concluded that a nonameric 9Y peptide (YVVPGSPCL) was the epitope for the Q1 TIL clone. Furthermore, the Q1 TIL clone did not recognize wild-type 9S peptide (SVVPGSPCL) (Fig. 4b). Therefore, the epitope for the Q1 TIL clone was the 9Y peptide (YVVPGSPCL), and the Q1 TIL clone specifically recognized this neoantigen. Furthermore, the other 21 TIL clones specific for 1226 RCC cells also recognized the 9Y peptide pulsed onto 293T-C*03:03 cells (data not shown), indicating that the 9Y peptide is a dominant neoantigen.

Identification of the epitope of the Q1 TIL clone. a Q1 TIL clone reactivity to serially truncated peptides. Serially truncated mutant peptides encoded by mutant TNS1 were examined for reactivity to the Q1 TIL clone. Each peptide was pulsed onto 293 T-C*03:03 cells at different concentrations, and their recognition by the Q1 TIL clone was assessed by an IFNγ ELISPOT assay. IFNγ spots are shown as mean ± SEM (n = 3). b The Q1 TIL clone recognizes the mutant peptide, but not the wild-type peptide. Two peptides, 9Y and 9S derived from mutated and non-mutated TNS1 gene, respectively, were examined for reactivity to the Q1 TIL clone. Each peptide was pulsed onto 293 T-C*03:03 cells at different concentrations, and their recognition by the Q1 TIL clone was assessed by an IFNγ ELISPOT assay. IFNγ spots are shown as mean ± SEM (n = 3)
Since we identified the TNS1 gene mutation in 1226 RCC cells, we confirmed the gene mutation and Q1 TIL clone reactivity using primary cells. Single separated cells derived from the red and yellow tumors were used as target cells. An ELISPOT assay demonstrated that the Q1 TIL clone recognized only red tumor single cell suspensions in an HLA-restricted manner, which was inhibited by an anti-HLA-BC mAb (Fig. 5a). The TNS1 gene in 1226 RCC cells, red tumor, yellow tumor, normal kidney, and PBMCs was amplified using PCR and the nucleotide substitutions in each sample were verified with DNA sequencing. We confirmed that the TNS1 mutation was present only in the red tumor (Fig. 5b).

Immunological and genetic heterogeneity of the primary tumor. a The Q1 TIL clone recognizes primary RCC cells derived from the red tumor, but not RCC cells derived from the yellow tumor. Q1 TIL clone reactivity for primary RCC cells derived from the red and yellow tumors. To confirm specificity, an anti-HLA-BC antibody (B1.23.2) was used. IFNγ spots are shown as mean ± SEM (n = 3). b TNS1 gene mutation is detected only in RCC cells derived from the red tumor. DNA sequencing was performed using genomic DNA derived from 1226 RCC cells, red tumor, yellow tumor, normal kidney, and PBMCs. The 1309Y mutation was detected only in 1226 RCC cells and the red tumor
Detection of Q1 TCR genes in a tumor sample, but not in peripheral blood
We analyzed the TCR repertoire in tumor samples, and identified TCR α-chain (TCRA) and β-chain (TCRB) sequences from the Q1 TIL clone using 5′-RACE (Supplementary Table 3). The inverse Simpson index (1/λ) was 541 and 396 for TCRA and TCRB in PBMCs and 232 and 155 in red tumor TILs, respectively (Fig. 6). Subsequently, we used TCRA and TCRB deep sequencing to determine the relative frequencies of the identified Q1 TIL clonotypes in red tumor TILs and PBMCs. TNS1 mutation-specific TCRA and TCRB were found in 1.08% and 0.68% of red tumor TILs, respectively, whereas neither was detected in PBMCs (Fig. 6).

Q1 TIL clone TCRs are detected in TILs, but not in PBMCs. The pie charts indicate the T-cell receptor alpha variable region (TRAV) and T-cell receptor beta variable region (TRBV) repertoires of PBMCs and TILs
Mutant TNS1 has a role in cell migration
Most neoantigens are encoded by passenger mutations, and only neoantigens derived from KRAS and TP53 mutations are encoded by driver mutations [44, 45]. Therefore, we evaluated whether the TNS1 mutation was functional. To address this, we constructed plasmids encoding the wild-type (1309S) or mutant (1309Y) TNS1 gene. The RCC Caki-1 and ACHN cell lines did not express TNS1 protein; thus, we over-expressed 1309S TNS1 and 1309Y TNS1 in each cell line. TNS1 protein expression was confirmed by western blot analysis (Fig. 7a). A previous study demonstrated that TNS1 has a role in cell migration [46]; therefore, we assessed cell migration using a wound healing assay. Over-expression of 1309S TNS1 did not increase cell migration, but over-expression of 1309Y TNS1 increased cell migration (Fig. 7b). Since a wound healing assay might be affected by the speed of cell growth, we examined the cell growth rate, but over-expression of 1309S and 1309Y did not affect cell growth (Fig. 7c). These results indicate that the TNS1 1309Y mutation has a role in cell migration, but not in cell growth.

Mutant TNS1 has a role in cell migration. a Over-expression of wild-type (1309S) and mutant (1309Y) TNS1. Wild-type and mutant TNS1 genes were constructed and stably transfected into the Caki-1 RCC cell line. Protein expression was assessed by western blot analysis. β-Actin was used as a positive control. b Mutant TNS1 has a role in cell migration. Cell migration was assessed in Caki-1 cells stably transfected with wild-type (1309S) or mutant (1309Y) TNS1 by a wound healing assay. The cells were cultured until confluent, then wounded with a pipette. Wound healing was observed at 24 h and 48 h later. Mock transfected cells were used as a negative control. c Mutant TNS1 does not have a role cell growth. Cell growth was assessed in Caki-1 cells stably transfected with wild-type (1309S) or mutant (1309Y) TNS1 by a cell count assay. The cells were seeded into 6-well plates, and cell numbers were counted after culture for 2 days. The data are shown as mean ± SEM (n = 3)
Discussion
In this study, we revealed that CD8+ CD137+ TILs recognize a 9-mer neoantigen, YVVPGSPCL, encoded by a missense mutation of the TNS1 gene. Although several TAAs of RCC have been identified previously, few reports have described tumor antigens of RCC recognized by CD8+ TILs, and the landscape of TAAs of RCC that are recognized by TILs is elusive [47,48,49,50,51]. TILs of RCC recognize neoantigens encoded by missense mutations [47, 51], encoded by an alternative open reading frame [48, 51], or produced by protein splicing [49, 50]. A number of over-expressed antigens and cancer-testis antigens have been described [21, 22, 52, 53], but no over-expressed or cancer-testis antigens have been described as being recognized by TILs of RCC. However, HLA ligandome analysis revealed that a number of TAAs are over-expressed in RCC tissue, and immunization against over-expressed antigens showed promising anti-tumor effects [54, 55]. Thus, neoantigens might be a powerful target, especially in immune checkpoint blockade, but other types of TAAs may also have roles in tumor rejection. A recent study revealed that melanoma patients treated with personalized neoantigen therapy showed a very long anti-tumor effect, and interestingly, epitope spreading was observed, suggesting that cancer-testis antigens may also have a role in the anti-tumor effect [56].
TCR repertoire analysis identified neoantigen-specific TCRA and TCRB that were recognized by approximately 1% of TILs, but were not detected in PBMCs. In most malignancies, the infiltration of CD8+ TILs predicts a better prognosis [57]; however, CD8+ TILs were proposed as a negative prognostic factor in RCC [30, 32]. This inconsistency suggested that either TILs of RCC could recognize tumor antigens or they were just bystander CD8+ TILs that are not specific for tumor antigens and could recognize a wide range of epitopes unrelated to cancer (such as those from EBV, human cytomegalovirus, or influenza virus) [34]. Tumor-specific TILs have been identified according to the expression of CD39 [34]. Other studies revealed that CD39+ and CD103+ TILs were specific for cancer cells [58]. These reports suggest that CD39 expression might be a good marker for tumor-specific TILs. However, recent studies on RCC revealed that high infiltration of CD39+ cells was correlated with a poorer prognosis [59, 60], suggesting that high infiltration rates of functional TILs might be correlated with poor prognosis in RCC. Interestingly, CD39+ CD103+ TILs have a distinct TCR repertoire compared with peripheral blood, suggesting that CD39+ CD103+ TILs might expand in the tumor site [58]. In this study, neoantigen-specific TCR was detected only in TILs, and not in PBMCs, suggesting that the neoantigen-specific CTLs expanded locally.
RCC with high predicted neoantigen load is associated with a better prognosis [61]. In this study, immunohistochemical staining revealed that CD8+ TILs were abundant in the neoantigen-positive high-grade RCC lesion, but immunosuppressive PD-1 expression was also higher in the same lesion, suggesting that functional TILs are suppressed by the PD-L1/PD-1 axis. These observations suggest that RCC is immunogenic with neoantigen expression due to a relative high tumor-mutational burden [62] or other types of neoantigens encoded by indel frameshifts or endogenous retroviruses [19, 20]. A previous interesting case report described that a RCC case after peptide vaccination showed significant response to PD-1 blockade. The histological analysis revealed that HLA-DR is expressed in TIL rich RCC area [63]. In this case HLA-DR is partially expressed in the red tumor (data not shown). Thus, PD-1 blockade might be effective in this case, unfortunately we could not treat due to high performance status (PS).
A histological and cytometrical approach revealed that higher tumor-grade was associated with higher number of TILs in clear cell RCC, as we observed in this study [64]. Interestingly, CD8 gene expression was not related to a better prognosis, but was correlated with the expression of genes for an immunosuppressive tumor microenvironment including cytotoxic T-lymphocyte antigen 4 (CTLA-4), PD-1, lymphocyte-activation gene 3 (LAG-3), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), PD-L1, PD-L2, indoleamine 2,3-dioxygenase 1, and IL-10 [61], suggesting that TILs induce a highly immunosuppressive tumor microenvironment, especially in RCC, which might abrogate the anti-tumor functions of TILs in RCC.
TNS1 is a 220-kDa protein that localizes to focal adhesions; it acts as a molecular bridge linking the extracellular matrix, actin cytoskeleton, and signal transduction [46, 65]. Although TNS1 is expressed in normal tissues, some studies suggested that its over-expression might be implicated in tumorigenesis in several types of tumor [66,67,68]. Recently, transgelin/TNS1 signaling was shown to promote cell proliferation and invasion in colorectal cancer, and that transgelin/TNS1 expression levels could potentially serve as a prognostic and therapeutic target in patients with colorectal cancer [69]. Other studies revealed that TNS1 expression was significantly elevated and the microRNA-152/TNS1 axis promotes the progression of non-small cell lung cancer [70]. These studies indicated that wild-type TNS1 is related to cell growth and invasion. In this study neither wild-type nor S1309Y TNS1 enhanced cell growth in ACHN RCC cells, but S1309Y TNS1 enhanced cell mobility compared with wild-type TNS1.
Phase 3 trials on RCC immunotherapy have revealed that treatment with an anti-PD-1, anti-PD-L1, or anti-CTLA-4 antibody improves the overall survival of patients with metastatic RCC, compared to molecular targeting therapies [9,10,11,12]. However, these immune checkpoint inhibitors, even with combination therapy, have shown complete responses in only 1–9% patients with metastatic RCC [9, 10, 12]. Although many negative regulators have been identified in cancer immunity, including PD-L1, LAG-3, and TIM-3, this inadequate efficacy of immunotherapy against RCC may be associated with tumor heterogeneity and branched tumor evolution [33, 71, 72]. Moreover, tumor gene heterogeneity may influence not only the malignant potential of a tumor but also its immune phenotype, due to differences in tumor mutational burden. In this study, we identified the tumor antigen driven from an intra-tumor heterogenic missense mutation of RCC that autologous TILs could recognize and use to infiltrate only the high-grade tumor. Of 2212 patients with RCC in TCGA data, 63 cases (2.8%) were identified to have TNS1 mutation, but S1309Y missense mutation were not detected in the database, suggesting TNS1 mutation is relative common in RCCs and may has role in RCCs.
To identify the epitopes of CTLs, HLA ligandome analysis, gene expression analysis, and reverse immunogenetics have been performed [73,74,75]. Actually, we previously performed HLA ligandome analysis using 1226 RCC cells [73]; however, we could not identify any CTL epitope. To overcome this, we used TMGs and identified a TNS1 gene mutation as a novel neoantigen. The TMG method is useful for detecting neoantigens encoded by missense or indel mutations [76, 77]; however, over-expressed antigens cannot be identified with TMGs. Thus, technological improvements are needed to identify orphan antigens recognized by CTLs. A recent approach using a yeast library enabled comprehensive antigenic peptide screening [78]. These technical innovations might speed up clinical trials using TCR-T cells encoded by clonal TILs.
In summary, we identified an RCC neoantigen that was recognized by autologous TILs using a modified TMG approach. We found a 9-mer epitope generated by a TNS1 missense mutation that was expressed with intra-tumor heterogeneity in only the high-grade red tumor. TILs could be isolated from only the TNS1 gene mutation-positive tumor. A TCR specific for the antigenic peptide could only be identified in the TNS1 gene mutation-positive tumor. These results suggest that neoantigen-specific TILs recognized RCC cells and infiltrated the antigen-positive lesion. These observations provide a novel insight into the anti-tumor effects of TILs.
References
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70:7–30
Fisher R, Gore M, Larkin J (2013) Current and future systemic treatments for renal cell carcinoma. Semin Cancer Biol 23:38–45
Ferlay J, Soerjomataram I, Dikshit R et al (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386
Hudes G, Carducci M, Tomczak P et al (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356:2271–2281
Motzer RJ, Escudier B, Oudard S et al (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372:449–456
Motzer RJ, Rini BI, Bukowski RM et al (2006) Sunitinib in patients with metastatic renal cell carcinoma. JAMA 295:2516–2524
Rini BI, Escudier B, Tomczak P et al (2011) Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 378:1931–1939
Sternberg CN, Davis ID, Mardiak J et al (2010) Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 28:1061–1068
Motzer RJ, Escudier B, McDermott DF et al (2015) Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 373:1803–1813
Motzer RJ, Tannir NM, McDermott DF et al (2018) Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 378:1277–1290
Motzer RJ, Penkov K, Haanen J et al (2019) Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 380:1103–1115
Rini BI, Plimack ER, Stus V et al (2019) Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 380:1116–1127
Motzer RJ, Bacik J, Murphy BA et al (2002) Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 20:289–296
Klapper JA, Downey SG, Smith FO et al (2008) High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma : a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer 113:293–301
Yang JC, Childs R (2006) Immunotherapy for renal cell cancer. J Clin Oncol 24:5576–5583
Brahmer JR, Tykodi SS, Chow LQM et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465
Lawrence MS, Stojanov P, Polak P et al (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218
Yarchoan M, Hopkins A, Jaffee EM (2017) Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 377:2500–2501
Turajlic S, Litchfield K, Xu H et al (2017) Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 18:1009–1021
Smith CC, Beckermann KE, Bortone DS et al (2018) Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest 128:4804–4820
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T (2014) Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 14:135–146
Sato E, Torigoe T, Hirohashi Y et al (2008) Identification of an immunogenic CTL epitope of HIFPH3 for immunotherapy of renal cell carcinoma. Clin Cancer Res 14:6916–6923
Gaugler B, Brouwenstijn N, Vantomme V et al (1996) A new gene coding for an antigen recognized by autologous cytolytic T lymphocytes on a human renal carcinoma. Immunogenetics 44:323–330
Brändle D, Brasseur F, Weynants P et al (1996) A mutated HLA-A2 molecule recognized by autologous cytotoxic T lymphocytes on a human renal cell carcinoma. J Exp Med 183:2501–2508
Van den Eynde BJ, Gaugler B, Probst-Kepper M et al (1999) A new antigen recognized by cytolytic T lymphocytes on a human kidney tumor results from reverse strand transcription. J Exp Med 190:1793–1800
Morel S, Lévy F, Burlet-Schiltz O et al (2000) Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity 12:107–117
Clemente CG, Mihm MC Jr, Bufalino R et al (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77:1303–1310
Dieci MV, Mathieu MC, Guarneri V et al (2015) Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann Oncol 26:1698–1704
Geng Y, Shao Y, He W et al (2015) Prognostic role of tumor-infiltrating lymphocytes in lung cancer: a meta-analysis. Cell Physiol Biochem 37:1560–1571
Badalamenti G, Fanale D, Incorvaia L et al (2019) Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone? Cell Immunol 343:103753
Nakano O, Sato M, Naito Y et al (2001) Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res 61:5132–5136
Remark R, Alifano M, Cremer I et al (2013) Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res 19:4079–4091
Giraldo NA, Becht E, Pagès F et al (2015) Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res 21:3031–3040
Simoni Y, Becht E, Fehlings M et al (2018) Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557:575–579
Morita R, Hirohashi Y, Nakatsugawa M et al (2014) Production of multiple CTL epitopes from multiple tumor-associated antigens. Methods Mol Biol 1139:345–355
Asano T, Hirohashi Y, Torigoe T et al (2016) Brother of the regulator of the imprinted site (BORIS) variant subfamily 6 is involved in cervical cancer stemness and can be a target of immunotherapy. Oncotarget 7:11223–11237
Akatsuka Y, Goldberg TA, Kondo E et al (2002) Efficient cloning and expression of HLA class I cDNA in human B-lymphoblastoid cell lines. Tissue Antigens 59:502–511
Nakatsugawa M, Hirohashi Y, Torigoe T et al (2009) Novel spliced form of a lens protein as a novel lung cancer antigen, Lengsin splicing variant 4. Cancer Sci 100:1485–1493
Morita S, Kojima T, Kitamura T (2000) Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7:1063–1066
Ogawa T, Hirohashi Y, Murai A et al (2017) ST6GALNAC1 plays important roles in enhancing cancer stem phenotypes of colorectal cancer via the Akt pathway. Oncotarget 8:112550–112564
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26:589–595
do Valle ÍF, Giampieri E, Simonetti G, et al (2016) Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinform 17:341
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Tran E, Robbins PF, Lu Y-C et al (2016) T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med 375:2255–2262
Deniger DC, Pasetto A, Robbins PF et al (2018) T-cell responses to TP53 “hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res 24:5562–5573
Chen H, Duncan IC, Bozorgchami H, Lo SH (2002) Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc Natl Acad Sci USA 99:733–738
Gaudin C, Kremer F, Angevin E et al (1999) A hsp70-2 mutation recognized by CTL on a human renal cell carcinoma. J Immunol 162:1730–1738
Ronsin C, Chung-Scott V, Poullion I et al (1999) A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J Immunol 163:483–490
Hanada K, Perry-Lalley DM, Ohnmacht GA et al (2001) Identification of fibroblast growth factor-5 as an overexpressed antigen in multiple human adenocarcinomas. Cancer Res 61:5511–5516
Hanada K-I, Yewdell JW, Yang JC (2004) Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 427:252–256
Hansen UK, Ramskov S, Bjerregaard A-M et al (2020) Tumor-infiltrating T cells from clear cell renal cell carcinoma patients recognize neoepitopes derived from point and frameshift mutations. Front Immunol 11:373
Vissers JL, De Vries IJ, Schreurs MW et al (1999) The renal cell carcinoma-associated antigen G250 encodes a human leukocyte antigen (HLA)-A2.1-restricted epitope recognized by cytotoxic T lymphocytes. Cancer Res 59:5554–5559
Nishizawa S, Hirohashi Y, Torigoe T et al (2012) HSP DNAJB8 controls tumor-initiating ability in renal cancer stem-like cells. Cancer Res 72:2844–2854
Flad T, Spengler B, Kalbacher H et al (1998) Direct identification of major histocompatibility complex class I-bound tumor-associated peptide antigens of a renal carcinoma cell line by a novel mass spectrometric method. Cancer Res 58:5803–5811
Walter S, Weinschenk T, Stenzl A et al (2012) Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 18:1254–1261
Hu Z, Leet DE, Allesøe RL et al (2021) Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. https://doi.org/10.1038/s41591-020-01206-4
Gooden MJM, de Bock GH, Leffers N et al (2011) The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer 105:93–103
Duhen T, Duhen R, Montler R et al (2018) Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9:2724
Qi Y, Xia Y, Lin Z et al (2020) Tumor-infiltrating CD39+ CD8+ T cells determine poor prognosis and immune evasion in clear cell renal cell carcinoma patients. Cancer Immunol Immunother 69:1565–1576
Wu J, Wang Y-C, Xu W-H et al (2020) High expression of CD39 is associated with poor prognosis and immune infiltrates in clear cell renal cell carcinoma. Onco Targets Ther 13:10453–10464
Matsushita H, Sato Y, Karasaki T et al (2016) Neoantigen load, antigen presentation machinery, and immune signatures determine prognosis in clear cell renal cell carcinoma. Cancer Immunol Res 4:463–471
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69–74
Kurahashi R, Motoshima T, Fukushima Y et al (2020) Remarkable antitumor effect of nivolumab in a patient with metastatic renal cell carcinoma previously treated with a peptide-based vaccine. IJU Case Rep 3:44–48
Kawashima A, Kanazawa T, Kidani Y et al (2020) Tumour grade significantly correlates with total dysfunction of tumour tissue-infiltrating lymphocytes in renal cell carcinoma. Sci Rep 10:6220
Lo SH, An Q, Bao S et al (1994) Molecular cloning of chick cardiac muscle tensin. Full-length cDNA sequence, expression, and characterization. J Biol Chem 269:22310–22319
Chen H, Ishii A, Wong W-K et al (2000) Molecular characterization of human tensin. Biochem J 351:403
Hall EH, Daugherty AE, Choi CK et al (2009) Tensin1 requires protein phosphatase-1alpha in addition to RhoGAP DLC-1 to control cell polarization, migration, and invasion. J Biol Chem 284:34713–34722
Kiflemariam S, Ljungström V, Pontén F, Sjöblom T (2015) Tumor vessel up-regulation of INSR revealed by single-cell expression analysis of the tyrosine kinome and phosphatome in human cancers. Am J Pathol 185:1600–1609
Zhou H, Zhang Y, Wu L et al (2018) Elevated transgelin/TNS1 expression is a potential biomarker in human colorectal cancer. Oncotarget 9:1107–1113
Duan J, Wang L, Shang L et al (2020) miR-152/TNS1 axis promotes non-small cell lung cancer progression through Akt/mTOR/RhoA pathway. Biosci Rep. https://doi.org/10.1042/BSR20201539
Gerlinger M, Rowan AJ, Horswell S et al (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366:883–892
Kawashima A, Kanazawa T, Goto K et al (2018) Immunological classification of renal cell carcinoma patients based on phenotypic analysis of immune check-point molecules. Cancer Immunol Immunother 67:113–125
Kochin V, Kanaseki T, Tokita S et al (2017) HLA-A24 ligandome analysis of colon and lung cancer cells identifies a novel cancer-testis antigen and a neoantigen that elicits specific and strong CTL responses. Oncoimmunology 6:e1293214
Tsukahara T, Nabeta Y, Kawaguchi S et al (2004) Identification of human autologous cytotoxic T-lymphocyte-defined osteosarcoma gene that encodes a transcriptional regulator, papillomavirus binding factor. Cancer Res 64:5442–5448
Hirohashi Y, Torigoe T, Maeda A et al (2002) An HLA-A24-restricted cytotoxic T lymphocyte epitope of a tumor-associated protein, survivin. Clin Cancer Res 8:1731–1739
Robbins PF, Lu Y-C, El-Gamil M et al (2013) Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19:747–752
Lu Y-C, Yao X, Crystal JS et al (2014) Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 20:3401–3410
Gee MH, Han A, Lofgren SM et al (2018) Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell 172:549-563.e16
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS), KAKENHI for T. Torigoe (17H01540) and Y. Hirohashi (20H03460). This work was also supported by the Japan Agency for Medical Research and Development (AMED), the Project for Cancer Research and Therapeutic Evolution (P-CREATE) for T. Torigoe (16770510) and T. Kanaseki (20cm0106352h0002), and the Japan Science and Technology Agency (JST), CREST Grant Number JPMJCR15G3 for S. Hashimoto.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Masahiro Matsuki, Yoshihiko Hirohashi, Munehide Nakatsugara, Aiko Murai, Terufumi Kubo, Shinichi Hashimoto, Serina Tokita, Kenji Murata, Takayuki Kanaseki, Tomohide Tsukahara, Sachiyo Nishida, Toshiaki Tanaka, Hiroshi Kitamura, Naoya Masumori and Toshihiko Torigoe. The first draft of the manuscript was written by Masahiro Matsuki, Yoshihiko Hirohashi, Naoya Masumori and Toshihiko Torigoe, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no financial conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Matsuki, M., Hirohashi, Y., Nakatsugawa, M. et al. Tumor-infiltrating CD8+ T cells recognize a heterogeneously expressed functional neoantigen in clear cell renal cell carcinoma. Cancer Immunol Immunother 71, 905–918 (2022). https://doi.org/10.1007/s00262-021-03048-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-021-03048-6