
Abstract
Introduction
The human papillomavirus (HPV) encoded oncoproteins E6 and E7 are constitutively expressed in HPV-associated cancers, making them logical therapeutic targets. Intramuscular immunization of patients with HPV16 L2E7E6 fusion protein vaccine (TA-CIN) is well tolerated and induces HPV-specific cellular immune responses. Efficacy of PD-1 immune checkpoint blockade correlates with the level of tumor-infiltrating CD8 + T cells, yet most patients lack significant tumor infiltration of immune cells making immune checkpoint blockade suboptimal. We hypothesized that intratumoral vaccination with TA-CIN could increase the number of tumor-infiltrating CD8 + T cells, synergize with PD-1 blockade and result in better control of tumors compared with either PD-1 blockade or vaccination alone.
Methods
We examined the immunogenicity and antitumor effects of intratumoral vaccination with TA-CIN alone or in combination with PD-1 blockade in the TC-1 syngeneic murine tumor model expressing HPV16 E6/E7.
Results
Intratumoral vaccination with TA-CIN induced stronger antigen-specific CD8 + T cell responses and antitumor effects. Intratumoral TA-CIN vaccination generated a systemic immune response that was able to control distal TC-1 tumors. Furthermore, intratumoral TA-CIN vaccination induced tumor infiltration of antigen-specific CD8 + T cells. Knockout of Batf3 abolished antigen-specific CD8 + T cell responses and antitumor effects of intratumoral TA-CIN vaccination. Finally, PD-1 blockade synergizes with intratumoral TA-CIN vaccination resulting in significantly enhanced antigen-specific CD8 + T cell responses and complete regression of tumors, whereas either alone failed to control established TC-1 tumor.
Conclusions
Our results provide rationale for future clinical testing of intratumoral TA-CIN vaccination in combination with PD-1 blockade for the control of HPV16-associated tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High-risk human papillomavirus (hrHPV) infection is responsible for 5% of all cancer cases globally [1], including 99% of all cervical cancers [2]. Approximately 50% of cervical cancer is related to HPV16 and about 20% with HPV18 [2]. In addition, a subset of anogenital and oropharyngeal malignancies in men and women is also associated with hrHPV, and these cancers are predominantly driven by HPV16. As persistent hrHPV infection is necessary for cancer development, cytologic screening and HPV testing have diminished the burden of cervical cancer in developed countries by approximately 80% [3]. The efficacy and safety of HPV L1 VLP immunization to prevent new HPV16 and HPV18 infections are well-proven [4]. However, for patients with persistent HPV infection or established HPV-associated cervical dysplasia, these preventive vaccines do not cause clearance [5]. Furthermore, conventional chemoradiation therapy for invasive HPV-associated cancers provides only limited benefit to patients, with a 30% five-year survival rate for patients with advanced cervical cancer [6]. As a result, there is a clear need for targeted treatment strategies, such as a therapeutic HPV vaccination, to clear HPV16 and other hrHPV infections and their associated diseases.
There are several types of therapeutic HPV vaccines that are currently under development [7]. Among these therapeutic vaccines is a protein based vaccine called Tissue Antigen—Cervical Intraepithelial Neoplasia (TA-CIN), which is a recombinant fusion protein consisting of two HPV16 oncoproteins, E6 and E7, as well as the minor capsid protein L2 [8]. TA-CIN is purified from E. coli and is administered in the form of a filterable aggregate, a form that potentially reduces its diffusion beyond the injection site and increases its uptake by phagocytes, such as antigen-presenting cells for cross-presentation. HPV encoded oncoproteins E6 and E7 are potential targets for immunotherapy against HPV-associated malignancies because they are constantly expressed in all HPV-associated cancer cells, are functionally required for the initiation and maintenance of disease, and, as foreign antigens, they are not subject to central immune tolerance [9]. The minor capsid protein L2 is a potential prophylactic antigen for HPV-associated precursor lesions and contains neutralizing epitopes to induce antibody response against a wide range of papillomavirus types [10, 11]. A phase I trial has demonstrated that serial intramuscular vaccinations with TA-CIN in the absence of adjuvant can generate HPV antigen-specific antibody and T-cell responses without any significant adverse effects [12]. Two phase II trials have investigated TA-CIN; one trial investigated TA-CIN protein as a booster vaccine administered after either recombinant HPV16/18 E6/E7 vaccinia virus (TA-HPV) or topical imiquimod administration, and the other trial used TA-CIN as a priming vaccination prior to the administration of TA-HPV [13, 14]. In the current study, we use TA-CIN.
Several therapeutic HPV vaccines have been used with immune checkpoint blockade to enhance the beneficial effects of the vaccine [15, 16]. Immune checkpoint blockades are a form of immunotherapy that targets immune checkpoint molecules such as PD-1/PD-L1 and CTLA-4 (for review see [17]). There are already commercially available checkpoint inhibitors, such as nivolumab, pembrolizumab, and atezolizumab (fda.gov). However, checkpoint inhibitors do not always elicit strong responses, which has generated further interest in boosting the effectiveness of this treatment by using combination therapies. For instance, there have been studies regarding combination of different PD-1 and PD-L1 inhibitors with chemotherapy, radiotherapy, and other immunotargeting therapies. At the time of writing this article, clinicaltrials.gov contains over 800 clinical studies investigating combination therapies of a PD-1/PD-L1 blockade and other cancer treatments for conditions ranging from esophageal cancer, gastric cancer, lung cancer, brain metastasis, and more (clinicaltrials.gov). Several possibilities exist to explain why PD-1 and PD-L1 blockades lack efficacy without combination treatment. Notably, the efficacies of these PD-1/PD-L1 antibody immunotherapies correlate with the level of tumor-infiltrating CD8 + T cells [18]. However, most cancer patients do not have significant tumor infiltration of immune cells, especially CD8 + T cells [19]. Therefore, the antitumor response of immune checkpoint blockade may be suboptimal in the tumors of cancer patients that lack immune cell infiltration. Induction of tumor-infiltrating CD8 + T cells is likely critical for the efficacy of immune checkpoint blockade. One approach to induce immune cell tumor infiltration in patients with HPV-associated malignancies is vaccinating patients with a therapeutic HPV protein-based vaccine, TA-CIN, which may induce CD8 + tumor infiltration.
Therapeutic HPV vaccines potentially can be used in combination therapies with PD-1/PD-L1 blockade since the treatments generate therapeutic antitumor effects through different mechanisms. In 2018, pembrolizumab was approved for the treatment of recurrent or metastatic cervical cancers expressing PD-L1 [20]. HPV vaccines and immune checkpoint blockade have been tested preclinically by using a PD-1 blockade and a Listeria-based vaccine. After observing that an E6/E7-expressing preclinical tumor model, TC-1, upregulates PD-L1 upon Listeria-based vaccination, one study combined HPV vaccination with a PD-1 blocking antibody and found that the combination led to reduction in or complete regression of tumor growth [16]. One of the first clinical trials to test combination therapy (NCT02426892) in HPV-associated cancers used subcutaneously administered HPV16 peptide-based vaccine and nivolumab, a PD-1 checkpoint inhibitor [15]. Although the study showed encouraging results, the trial was overall inconclusive [15]. Our presented research builds upon these prior studies by exploring the potential of intratumoral injection rather than using subcutaneous or intramuscular administration.
In order to investigate possible techniques to maximize immune cell tumor infiltration, we included intratumoral injection as a potential injection method. Intratumoral injection of immunostimulatory or immunotherapeutic treatments is a fledgling approach that seeks to improve the efficacy of immunotherapeutic cancer methods while minimizing systemic harm by administering the vaccine locally. There has been some success with intratumoral injection of media such as PD-1 antibodies, plasmid DNA vaccines, viruses, and cytokines [21, 22]. Enhanced antitumor immune stimulatory responses have been observed after intratumoral peptide injection when compared to other potential routes, additionally noting that intratumoral injection may also promote cytotoxic T lymphocytes to better infiltrate solid tumors [23]. Intratumoral vaccination administration is a promising approach as it permits a strong immune response with minimal systemic toxicity, and the local administration builds an immune response that may even effect on distal tumors that were not injected directly [21].
In this study, we hypothesized that intratumoral vaccination with therapeutic HPV protein vaccine TA-CIN could significantly increase the number of tumor-infiltrating CD8 + T cells in mice injected with HPV16 E6/E7 expression murine tumor TC-1 cells [24] and thereby synergize with PD-1 blockade. By combining intratumoral injection, immune checkpoint blockade, and TA-CIN vaccination, we aim to elicit a significant immune response. Studying immune responses elicited by intratumoral versus intramuscular vaccination alongside checkpoint blockade inhibition will provide insights on the potential effects of intratumoral vaccination on HPV-malignancies that are currently understudied. We compared the T cell responses and antitumor effects between different antigens and vaccination routes, and we characterized the mechanistic basis of intratumoral TA-CIN vaccination. Finally, we demonstrated that administering intratumoral TA-CIN vaccination in combination with anti-PD-1 antibody resulted in better control of tumors compared with either PD-1 blockade or vaccination alone.
Materials and Methods
Mice
5–8-week-old female C57BL/6 mice were purchased from Taconic Biosciences (Germantown, NY). Batf3 knockout mice of the C57BL/6 background were purchased from Jackson Laboratories (Farmington, CT). All mice were maintained at the Johns Hopkins University School of Medicine (Baltimore, MD) animal facility under specific pathogen-free conditions. All procedures were performed under a prior-approved protocol of the Johns Hopkins Animal Care and Use Committee, and in accordance with recommendations for the proper use and care of laboratory animals.
Peptides, antibodies, and regents
HPV 16 E7aa49-57 peptide, RAHYNIVTF, and HPV 16 E7aa43-62 peptide, GQAEPDRAHYNIVTFCCKCD, were synthesized by GenScript (Piscataway, NJ). FITC-conjugated anti-mouse CD4 (clone RM4-5), FITC- and PerCP-conjugated anti-mouse CD8a (clone 53.6.7), APC-conjugated anti-mouse CD3 (clone 17A2), and PE- and APC-conjugated anti-mouse CD45 (clone 30-F11) antibodies were purchased from BD Pharmingen (San Diego, CA). FITC-conjugated anti-mouse Gr-1 (clone RB6-8C5), PE-conjugated anti-mouse CD11b (clone M1/70), anti-Foxp3 (clone FJK-16 s) were purchased from eBioscience (San Diego, CA). FITC-conjugated anti-gp38, APC-conjugated anti-mouse PD-1 (clone RMP1-30), and anti-mouse PD-L1 (clone 10F-9G2) antibodies were purchased from Biolegend (San Diego, CA). PE-conjugated, HPV 16 E7aa49-57 peptide-loaded H-2Db tetramers were purchased from MBL International (Japan). Recombinant mouse IFN-γ was purchased from eBioscience. Purified anti-mouse PD-1 monoclonal antibodies (clone 29F.1A12) were purchased from Bio X Cell (West Lebanon, NH).
Cell line
The establishment of HPV16 E6- and E7-expressing TC-1 cell line has been described previously [24]. The cells were maintained in RPMI-1640 media supplemented with 2 mM of glutamine, 1 mM of sodium pyruvate, 100 nM of non-essential amino acids, 100 IU/mL penicillin, 100 μg/mL streptomycin (all from gibco of Life Technologies, Grand Island, NY), and 10% fetal bovine serum (FBS, from HyClone, Logan, UT).
Vaccine and vaccination
The production of TA-CIN has been described previously [8]. TA-CIN is a fusion protein vaccine comprised of HPV16 viral proteins L2, E6, and E7 for the treatment of HPV16-associated cervical cancer [8]. For vaccination, TA-CIN and peptide were diluted in PBS as indicated concentration. 20 µL were injected either intratumorally or intramuscularly (biceps femoris muscle), as indicated.
In vitro IFN-γ treatment of TC-1 tumor cells and detection of PD-L1 expression
2 × 105 of TC-1 tumor cells were plated in 6-well plates. The cells were treated with 10 ng/mL of recombinant mouse IFN-γ overnight. PD-L1 expression was detected by staining the cells with APC-conjugated anti-mouse PD-L1 antibody followed by the acquisition with FACSCalibur flow cytometer and analyzed with CellQuest Pro software (BD biosciences, Mountain View, CA).
In vivo tumor treatment experiment
For in vivo tumor treatment experiments, female C57BL/6 mice (5 mice per group) were injected with 2 × 105 of TC-1 tumor cells subcutaneously. The tumor-bearing mice were vaccinated as indicated. As for treatment with anti-mouse PD-1 monoclonal antibodies, the mice were injected with purified anti-mouse PD-1 antibody at a dose of 200 μg/mouse in 100 μL in PBS via intraperitoneal injection three times per week. Tumor growth was monitored twice a week by palpation and digital caliper measurement. Tumor volume was calculated with the formula [largest diameter × (perpendicular diameter)2] × 3.14/6. Either a tumor diameter greater than 2 cm or a natural death were recorded as death to calculate the survival of the tumor-bearing mice.
Preparation of tumor-infiltrating immune cells from TC-1 tumors
TC-1 tumors were surgically resected and placed in RPMI-1640 media containing 100 U/mL penicillin and 100 μg/mL streptomycin, then washed with PBS to remove blood contamination. The tumors were then cut into small pieces and digested with serum-free RPMI-1640 media containing 0.05 mg/mL collagenase I, 0.05 mg/mL collagenase IV, 0.025 mg/mL hyaluronidase IV, 0.25 mg/mL DNase I, 100 U/mL penicillin, and 100 μg/mL streptomycin. The tissues were incubated at 37 °C for 1 h with periodic agitation. The digested tissues were then filtered through a 70-μM nylon cell strainer. The resultant tumor-infiltrating immune cells were washed twice in Hank’s buffered salt solution (HBSS) (400 g for 10 min), and viable cells were counted using trypan blue dye exclusion.
Flow cytometric analysis
For tetramer staining, cells were stained with 0.5 μg (1 μL) of purified anti-mouse CD16/32 (Fc block, BD Pharmingen, San Diego, CA) at room temperature for 5 min. The cells were subsequently stained with anti-mouse CD8-FITC and PE-conjugated H-2Db tetramers loaded with HPV 16 E7aa49-57 peptide at 4 °C or anti-mouse PD-1-APC, as indicated. Following washing, the cells were stained with 3 μL/sample of 7-Aminoactinomycin D (7-AAD) immediately before undergoing flow cytometry analysis to distinguish dead cells. For the detection of Tregs, cells were incubated with anti-CD4-FITC, anti-CD8-PerCP and CD3-APC antibodies for 30 min at 4 °C. The cells were then permeabilized and fixed as per manufacturer’s instructions (eBioscience, San Diego, CA). The cells were further stained with anti-FoxP3-PE antibody for 30 min at 4 °C. For detection, CD11b+Gr-1+ myeloid-derived suppressor cells (MDSCs) were stained with FITC-conjugated anti-mouse Gr-1, PE-conjugated anti-mouse CD11b antibodies and APC-conjugated anti-mouse CD45. To detect PD-L1 expression by TC-1 tumor cells harvested from in vivo tumors, the cells were stained with FITC-conjugated anti-gp38, PE-conjugated anti-mouse CD45, and APC-conjugated anti-mouse PD-L1 antibodies. Cells were acquired with a FACSCalibur flow cytometer (BD Biosciences, Mountain View, CA) and analyzed with CellQuest Pro software (BD Biosciences, Mountain View, CA).
Statistical analysis
Data were expressed as means ± standard deviations (SD). Comparisons between individual data points were analyzed by two-tailed Student’s t test with Prism software from GraphPad (San Diego, CA). Survival of the tumor-bearing mice was analyzed by Kaplan–Meier analysis with Prism software. A p value of < 0.05 was considered statistically significant.
Results
Intratumoral vaccination with TA-CIN generated significantly better HPV16 E7-specific CD8 + T cell responses and antitumor effects compared to intramuscular vaccination of mice bearing established TC-1 tumor
We aimed to determine whether the route of TA-CIN vaccination impacts the generation of antigen-specific CD8 + T cell responses and antitumor effects. To address this question, C57BL/6 mice (5 mice/group) were first challenged with 2 × 105 TC-1 cells subcutaneously, and subsequently vaccinated with TA-CIN (25 μg/mouse) via either intramuscular or intratumoral injection (Fig. 1a). One week after the final vaccination, peripheral blood mononuclear cells (PBMC) were collected and E7-specific CD8 + T cells responses were compared. Tumor volumes were measured twice a week and survival of tumor-bearing mice was recorded. As shown in Fig. 1b, mice vaccinated with TA-CIN via the intratumoral route displayed the highest percentage of E7-specific CD8 + T cells in peripheral blood. Furthermore, mice vaccinated with TA-CIN intratumorally exhibited significantly reduced tumor growth and extended survival when compared to either untreated mice or mice vaccinated intramuscularly (Fig. 1c, d). These results suggest that intratumoral vaccination generates comparatively better therapeutic antitumor immunity in mice.

Intratumoral vaccination of TA-CIN generated significantly better HPV 16 E7-specific CD8 + T cell responses and antitumor effects in the TC-1 tumor model. a Schematic illustration of the experiment. Briefly, 5–8-week-old female C57BL/6 mice (5 mice/group) were injected with 2 × 105 TC-1 cells subcutaneously on Day 0. Three days later, the TC-1 tumor-bearing mice were vaccinated with 25 μg/mouse of TA-CIN vaccine in 20 μL via either intramuscular or intratumoral injection. The mice were boosted twice with the same regimen in 3–4-day intervals. One week after the last vaccination, PBMCs were prepared and stained with anti-mouse CD8 and HPV 16 E7 tetramer. The data were acquired with FACSCalibur flow cytometer and analyzed with CellQuest. Tumors were measured twice a week and survival of the tumor-bearing mice was recorded. b Summary of the flow cytometry data. c Summary of TC-1 tumor volume. Statistical significance between TA-CIN (I.M.) and TA-CIN (I.T.) had a p value of 0.0085. Statistical significance between untreated and TA-CIN (I.T.) had a p value of < 0.0001. d Kaplan–Meier survival analysis of TC-1 tumor-bearing mice treated with the different regimens. Statistical significance between TA-CIN (I.M.) and TA-CIN (I.T.) had a p value of 0.0114. Statistical significance between untreated and TA-CIN (I.T.) had a p value of 0.0003
Intratumoral vaccination of TA-CIN generated significantly better HPV16 E7-specific CD8 + T cell responses and antitumor effects in the TC-1 tumor model compared to intratumoral vaccination with either HPV16 E7 short or long peptides
We tested whether the TA-CIN vaccine generates stronger E7-specific antitumor immunity compared to either the E7 short or long peptide vaccines alone [18]. C57BL/6 mice (5 mice/group) were first challenged with 2 × 105 TC-1 cells subcutaneously and subsequently intratumorally vaccinated with one of the following reagents: TA-CIN (6.25 μg/mouse), HPV16 E7 aa49-57 peptide (0.088 μg/mouse), or HPV16 E7 aa43-62 peptide (0.184 μg/mouse) (Fig. 2a). One week after the last vaccination, PBMCs were collected and E7-specific CD8 + T cells responses were compared. Tumor volumes were measured twice a week and mice were monitored for survival. As shown in Fig. 2b, mice vaccinated with TA-CIN protein elicited the highest percentage of E7-specific CD8 + T cells compared to E7 short or long peptides. Both E7aa49-57 (short peptide) and E7aa43-62 (long peptide) inhibited TC-1 tumor growth significantly and prolonged the survival of TC-1 tumor-bearing mice compared to untreated mice. However, intratumoral TA-CIN vaccination induced a significantly stronger anti-tumor effect and improved the survival duration of TC-1 tumor-bearing mice when compared to E7 short or long peptides (Fig. 2c, d).

Intratumoral vaccination of TA-CIN, but not HPV 16 E7 short or long peptide, generated significantly better HPV 16 E7-specific CD8 + T cell responses and antitumor effects in the TC-1 tumor model. a Schematic illustration of the experiment. Briefly, 5–8-week-old female C57BL/6 mice (5 mice/group) were injected with 2 × 105 TC-1 cells subcutaneously on Day 0. Four days later, the TC-1 tumor-bearing mice were vaccinated with equal amounts of 6.25 μg/mouse of TA-CIN, 0.088 μg/mouse of HPV 16 E7aa49-57 peptide, or 0.184 μg/mouse of HPV16 E7aa43-62 peptide in 20 μL via intratumoral injection. The mice were boosted twice with the same regimen in 3-day intervals. One week after the final vaccination, PBMCs were prepared and stained with anti-mouse CD8 and HPV 16 E7 tetramer. The data were acquired with FACSCalibur flow cytometer and analyzed with CellQuest. Tumors were measured twice a week and survival of the tumor-bearing mice was recorded. b Summary of the flow cytometry data. c Summary of TC-1 tumor volume. Statistical significance between E7aa49-57 peptide and untreated had a p value of 0.0004. Statistical significance between E7aa43-62 peptide and untreated had a p value of 0.0002 for the tumor volume at day 28. Statistical significance between TA-CIN and E7aa49-57 peptide had a p value of 0.0004 for the tumor volume at day 28. Statistical significance of TA-CIN and E7aa43-62 peptide had a p value of < 0.0001 for the tumor volume at day 28. d Kaplan–Meier survival analysis of TC-1 tumor-bearing mice treated with the different regimens. Statistical significance between both E7aa49-57 peptide and E7aa43-62 peptide treatment groups compared to untreated had p values of 0.0011. Statistical significance for both E7aa49-57 peptide and E7aa43-62 peptide treatment groups compared to TA-CIN had P values of 0.0002 p = 0.0002
Intratumoral vaccination of TA-CIN generated systemic antitumor responses that control the growth of distant tumors
Intratumoral immune therapies can potentially elicit therapeutic antitumor immunity not only in the injected tumor site but also in distant noninjected tumor lesions, termed an abscopal effect [25]. To test whether the intratumoral vaccination of TA-CIN is also able to generate distal antitumor immune responses, we subcutaneously challenged C57BL/6 mice (5 mice/group) with 2 × 105 TC-1 cells on both sides of the mouse abdomen. Once the tumor was established, mice received an intratumoral injection of TA-CIN (6.25 μg/mouse) on only one side (Fig. 3a). To monitor the antitumor responses, tumors on both the ipsilateral and contralateral sides were measured twice a week. We found that robust regression was observed on both the ipsilateral and contralateral tumors (Fig. 3b, c). These results suggest that intratumoral vaccination of TA-CIN is able to generate systemic antitumor responses to control distant tumor growth.

Intratumoral vaccination of TA-CIN generated systemic E7-specific CD8 + T cell responses that are able to control the growth of distant tumors. a Schematic illustration of the experiment. Briefly, groups of 5–8-week-old female C57BL/6 mice (5 mice/group) were injected with 2 × 105 TC-1 tumor cells were subcutaneously injected on both sides of the mouse abdomen. A total of 6.25 μg/mouse of TA-CIN was injected intratumorally into the tumor of one side (ipsilateral) three times in 3–4-day intervals. TC-1 tumor growth was monitored on both the ipsilateral and contralateral sides. b Tumor growth curve of ipsilateral TC-1 tumors. c Tumor growth curve of contralateral TC-1 tumors. p < 0.0001 for both ipsilateral and contralateral tumor treated with TA-CIN when compared to untreated on day 25
Intratumoral injection of TA-CIN vaccination led to higher levels of E7-specific CD8 + T cells, and lower levels of regulatory T cells and MDSCs than intramuscular injections in both peripheral blood and local tumors
We have demonstrated that intratumoral injection of the TA-CIN vaccine induced stronger antitumor immunity than either intramuscular injection or untreated controls (Fig. 1), and is able to generate systemic antitumor effects (Fig. 3). To test whether the systemic and local immune profiles are consistent with the difference in antitumor effects, we compared the systemic and tumor-infiltrating E7-specific CD8 + T cells, regulatory T cells, and MDSCs after either intramuscular or intratumoral TA-CIN vaccination. C57BL/6 mice (5 mice/group) were first subcutaneously challenged with TC-1 tumor cells and subsequently vaccinated with the TA-CIN vaccine (6.25 μg/mouse) via either intramuscular or intratumoral injection on day 7. The mice were boosted twice with the same regimen at 3-day intervals. One week after the final vaccination, PBMCs and tumor-infiltrating immune cells were prepared in order to compare levels of E7-specific CD8 + T cells, regulatory T cells, and MDSCs. As shown in Fig. 4a–c, mice intratumorally vaccinated with TA-CIN generated the highest levels of E7-specific CD8 + T cells and the lowest levels of regulatory T cells and MDSCs in peripheral blood compared to either intramuscularly vaccinated mice or untreated controls. Furthermore, intratumoral TA-CIN vaccination also led to the highest levels of total CD8 + T cells and E7-specific CD8 + T cells, and the lowest levels of regulatory T cells in local tumors (Fig. 4d–f). Taken together, our data suggest that the local and systemic immune profiles of intratumoral TA-CIN vaccination are consistent with stronger local and distant antitumor immunity, as indicated by higher levels of E7-specific CD8 + T cells and lower levels of suppressive immune cells in the local tumors and peripheral blood.

Comparison of systemic and tumor-infiltrating E7-specific CD8 + T cells, regulatory T cells, and myeloid-derived suppressor cells after either intramuscular or intratumoral TA-CIN vaccination Groups of 5–8-week-old female C57BL/6 mice (5 mice/group) were vaccinated with 6.25 μg/mouse of TA-CIN via either intramuscular or intratumoral injection on Day 7 after TC-1 tumor cells were subcutaneously injected. The mice were boosted twice with the same regimen in 3-day intervals. One week after the final vaccination, PBMCs and tumor-infiltrating immune cells were prepared, and E7-specific CD8 + T cells, regulatory T cells, and myeloid-derived suppressor cells were analyzed by flow cytometry analysis. The data were acquired with FACSCalibur flow cytometer and analyzed with CellQuest. a E7-specific CD8 + T cells in peripheral blood. b Regulatory T cells in peripheral blood. c Myeloid-derived suppressor cells in peripheral blood. d Total number of tumor-infiltrating CD8 + T cells. e Number of tumor-infiltrating E7-specific CD8 + T cells. f Frequency of tumor-infiltrating regulatory T cells
Robust E7-specific CD8 + T cell induction and antitumor immunity after intratumoral TA-CIN vaccination is dependent on a Batf3-regulated pathway
Batf3 is an important pathway for the cross-presentation of tumor antigens to dendritic cells (DCs) and is required for T cell dependent immune therapies [26]. To examine whether the induction of E7-specific CD8 + T cells and antitumor immunity after intratumoral TA-CIN vaccination is dependent on the Batf3 pathway, we subcutaneously injected wild-type and Batf3 knockout C57BL/6 mice (5 mice/group) with 2 × 105 TC-1 cells and subsequently vaccinated them with TA-CIN (6.25 μg/mouse) via intratumoral injection (Fig. 5a). One week after the final vaccination, PBMCs were prepared and E7-specific CD8 T + cell responses were compared. The tumors were measured twice a week and survival of tumor-bearing mice was recorded. As shown in Fig. 5b, wild-type mice injected intratumorally with the TA-CIN vaccine generated a significant E7-specific CD8 + T cell response, while Batf3 knockout mice showed a significant decrease in T cell response. Moreover, Batf3 knockout completely abolished the antitumor effects in mice intratumorally vaccinated with TA-CIN, resulting in decreased survival (Fig. 5c, d). These results suggest that the Batf3-regulated pathway is required for E7-specific CD8 + T cell induction and generation of antitumor immunity after intratumoral TA-CIN vaccination.

Robust E7-specific CD8 + T cell induction and antitumor immunity after intratumoral TA-CIN vaccination is dependent on the Batf3-regulated pathway. a Schematic illustration of the experiment. Groups of 5–8-week-old female wild type and Batf3-knockout C57BL/6 mice (5 mice/group) were injected with 2 × 105 TC-1 cells subcutaneously on Day 0. Seven days later, the TC-1 tumor-bearing mice were vaccinated with 6.25 μg/mouse of TA-CIN vaccine in 20 μL via intratumoral injection. The mice were boosted twice with the same regimen in 3-day intervals. One week after the final vaccination, PBMCs were prepared and stained with anti-mouse CD8 and HPV 16 E7 tetramer. The data were acquired with FACSCalibur flow cytometer and analyzed with CellQuest. Tumors were measured twice a week and survival of the tumor-bearing mice was recorded. b Summary of the flow cytometry data. c Summary of TC-1 tumor volume. Statistical significance between treated and untreated Batf3 knockout mice had a p value of 0.7313. Statistical significance between treated Batf3 knockout and treated wild-type mice had a p value of < 0.0001 on day 28). d Kaplan–Meier survival analysis of TC-1 tumor-bearing mice treated with the different regimens. Statistical significance between TA-CIN treated and untreated knockout mice had a p value of 0.5485. Statistical significance between treated Baft3 knockout and treated wild-type mice had a p value of 0.0002
Intratumoral TA-CIN vaccination increased expression of PD-L1 by TC-1 tumor cells and expression of PD-1 by TC-1 tumor-infiltrating E7-specific CD8 + T cells
Many studies have shown that the PD-1/PD-L1 axis is the major mechanism for the functional inhibition of T cells at the site of tumors [27]. We wanted to determine why intratumoral injection of TA-CIN did not completely eliminate the tumors [28]. Previous study has demonstrated that vaccination can upregulate PD-L1 expression in the tumor microenvironment [29]. Therefore, we examined whether after intratumoral TA-CIN vaccination the tumor cells and tumor-infiltrating E7-specific CD8 + T cells expressed higher levels of PD-L1 and PD-1. C57BL/6 mice were first subcutaneously challenged with 2 × 105 TC-1 cells, and subsequently vaccinated with TA-CIN (6.25 μg/mouse) via intratumoral injection or left untreated. The mice in the vaccinated group were boosted with the same regimen twice in 3-day intervals. One week after the final vaccination, single cells were prepared from the excised TC-1 tumors for comparison of PD-1 and PD-L1 expression. As shown in Fig. 6b, TC-1 tumor cells from TA-CIN-vaccinated mice, which were determined as CD45– and gp38 + , expressed higher levels of PD-L1 when compared to TC-1 cells from either the untreated mice or isotype control. This increased PD-L1 expression after in vivo intratumoral TA-CIN vaccination was comparable to IFNγ treatment of TC-1 tumor cells in vitro (Fig. 6a, b). Staining of the tumor cells indicated that vaccination up-regulated PD-L1 expression by the tumor cells. Furthermore, the tumor-infiltrating CD8 + T cells from mice intratumorally vaccinated with TA-CIN, which were over 50% E7-positive (Fig. 6c), expressed higher PD-1 levels than tumor-infiltrating CD8 + T cells from untreated mice (Fig. 6d). Collectively, our data indicate that intratumoral TA-CIN vaccination increases PD-L1 expressions by TC-1 tumor cells and PD-1 expression by TC-1 tumor-infiltrating E7-specific CD8 + T cells. These results are likely to be a strategy for local tumors to counter the induction of tumor-infiltrating CD8 + T cells induced by intratumoral TA-CIN vaccination.

Analysis of PD-L1 expression by TC-1 cells cultured in vitro or isolated from tumors in vivo and PD-1 expression by TC-1 tumor-infiltrating HPV 16 E7-specific CD8 + T cells. a Flow cytometry analysis of PD-L1 expression by TC-1 cells cultured in vitro. Briefly, 1 × 105 TC-1 cells were plated in each well of a 6-well plate, then treated with 2.5 ng/mL of recombinant mouse IFN-γ for 48 h or left untreated. The cells were then stained with PE-conjugated anti-mouse PD-L1 (clone 10F.9G2) antibody or isotype control. The cells were acquired with FACSCalibur and analyzed with CellQuest Pro software. b PD-L1 expression by TC-1 cells from transplanted tumor. 6–8-week-old female C57BL/6 mice were injected with 2 × 105 TC-1 cells subcutaneously on Day 0. On Day 7, one group of tumor-bearing mice were vaccinated with 6.25 μg/mouse of TA-CIN (in 20 μL) through intratumoral injection. The mice were boosted with the same regimen twice in 3-day intervals. Another group of TC-1 tumor-bearing mice was left untreated. 7 days after the final vaccination, TC-1 tumors were excised, minced, and enzymatically digested. The resulting single-cell preparation was stained with either FITC-conjugated anti-gp38, PE-conjugated anti-mouse CD45, or APC-conjugated anti-mouse PD-L1 antibodies, as well as isotype controls. The cells were acquired with FACSCalibur and analyzed with CellQuest Pro software. TC-1 tumor cells were determined as CD45-negative and gp38-positive. Detection of frequency of (c) HPV 16 E7-specific CD8 + T cells from TC-1 tumor-infiltrating lymphocytes and (d) expression of PD-1 by HPV 16 E7-specific CD8 + T cells from TC-1 tumor-infiltrating lymphocytes. As described in (b), the resulting single-cell preparation from TC-1 tumors was stained with either FITC-conjugated anti-mouse CD8a, PE-conjugated HPV 16 E7aa49-57 peptide loaded H-2Db tetramer, or APC-conjugated anti-mouse PD-1 antibodies, as well as isotype controls. The cells were acquired with FACSCalibur and analyzed with CellQuest Pro software
Anti-PD-1 antibodies synergize with intratumoral TA-CIN vaccination to elicit complete regression of TC-1 tumors
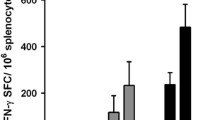
Since TC-1 tumor cells and tumor-infiltrating CD8 + T cells expressed higher levels of PD-L1 and PD-1 after intratumoral TA-CIN vaccination, we hypothesized that the addition of anti-mouse PD-1 treatment would enhance the immunogenicity and antitumor effects of intratumoral TA-CIN vaccine. To test this hypothesis, we subcutaneously injected C57BL/6 mice (5 mice/group) with 2 × 105 TC-1 cells and subsequently treated them with either anti-mouse PD-1 antibodies (200 μg/mouse) through intraperitoneal injection alone, TA-CIN (6.25 μg/mouse) through intratumoral injection alone, or administered both treatments. Anti-mouse PD-1 antibodies were given three times a week, and TA-CIN was boosted twice (Fig. 7a). One week after the final vaccination, PBMCs were prepared and E7-specific CD8 + T cell responses were compared. The tumors were measured twice a week and survival of the tumor-bearing mice was recorded. As shown in Fig. 7b, mice injected intratumorally with TA-CIN and anti-mouse PD-1 antibodies generated the highest E7-specific CD8 + T cell response, compared to either treatment alone. Furthermore, co-treatment of intratumoral TA-CIN vaccination together with anti-mouse PD-1 antibodies led to the complete inhibition of tumor growth as well as a 100% survival rate in TC-1 tumor-challenged mice, while either intratumoral TA-CIN vaccination or anti-mouse PD-1 antibody administration alone could only generate partial antitumor effects and resulted in only 20% and 0% survival of TC-1 tumor-challenged mice at day 120, respectively (Fig. 7c, d). Taken together, these results demonstrate that anti-PD-1 antibodies synergize with intratumoral vaccination of TA-CIN, resulting in significantly enhanced E7-specific CD8 + T cell responses and the complete regression of TC-1 tumors compared to either anti-PD-1 antibody administration or TA-CIN vaccination alone.

Comparison of HPV 16 E7-specific CD8 + T cell responses and anti-tumor effects of the TC-1 tumor-bearing mice after vaccination with TA-CIN through intratumoral injection with or without anti-mouse PD-1 treatment. a Schematic illustration of the experiment. Briefly, 6–8-week-old female C57BL/6 mice (5 mice/group) were injected with 2 × 105 TC-1 cells subcutaneously on Day 0. On Day 3, tumor-bearing mice were treated with either anti-mouse PD-1 antibody (200 μg/mouse) through intraperitoneal injection alone, vaccinated with 6.25 μg/mouse of TA-CIN (in 20 μL) through intratumoral injection alone, or treated with both. Anti-mouse PD-1 antibody was given three times a week, and TA-CIN was boosted twice as indicated. 7 days after the final vaccination, PBMCs were prepared from the mice, and stained with purified anti-mouse CD16/32 antibody first. The cells were then stained with FITC-conjugated anti-mouse CD8a antibody and PE-conjugated HPV 16 E7aa49-57 peptide-loaded H-2Db tetramer. The cells were acquired with FACSCalibur and analyzed with CellQuest Pro software. The tumor growth was monitored twice a week with a digital caliper. The death of the mouse was recorded as either natural death or when tumor size exceeded 2 cm in diameter. b Summary of the flow cytometry analysis of HPV 16 E7-specific CD8 + T cells. c Tumor growth curve. d Kaplan–Meier survival analysis of TC-1 tumor-bearing mice
Discussion
In this study, we found that intratumoral vaccination with TA-CIN generated potent antitumor immunity. Mice receiving this treatment had significantly higher levels of E7-specific CD8 + T cells, inhibited tumor growth, and increased rate of survival during the study period compared to the mice who received TA-CIN intramuscularly. This pattern of higher E7-specific CD8 + T cells, inhibited tumor growth, and increased survival was reflected in intratumoral vaccination with TA-CIN when compared to intratumoral vaccination with E7 short or E7 long peptides. These results are likely due to the structure of TA-CIN protein that can facilitate internalization by antigen-presenting cells, such as dendritic cells. The additional immunogenicity from L2 of TA-CIN may also contribute to the improved E7-specific CD8 + T cell responses and antitumor effects [8, 10]. Therefore, intratumoral injection of TA-CIN may significantly boost antitumor immunity. Moreover, intratumoral TA-CIN vaccination generated systemic immune responses that could significantly reduce tumor volume of distant TC-1 tumors. We also observed that intratumoral TA-CIN vaccination induced significantly more systemic and tumor-infiltrating antigen-specific CD8 + T cells while reducing the number of regulatory T cells and MDSCs in local tumor and peripheral blood. Mechanistically, we found that the potent immunogenicity and antitumor effects of intratumoral TA-CIN vaccination are governed by the Batf3 pathway, as knocking out Batf3 abolished antigen-specific CD8 + T cell responses and antitumor effects. We discovered that intratumoral vaccination with TA-CIN increased PD-L1 expression by TC-1 tumor cells and PD-1 expression by TC-1 tumor-infiltrating CD8 + T cells. Finally, we have demonstrated that PD-1 blockade synergized with intratumoral TA-CIN vaccination resulting in significantly enhanced antigen-specific CD8 + T cell responses, a complete regression of tumors, and a 100% survival rate at day 120 when compared to either PD-1 administration or intratumoral vaccination alone.
Our approach to combining intratumoral TA-CIN vaccination with PD-L1 blockade to increase anti-tumor immunogenicity is a potentially promising strategy for future clinical translation, and this approach may be applicable to many HPV-associated malignancies. Our use of a subcutaneous TC-1 tumor model results in murine tumors that are close to body surfaces and readily available to intratumoral vaccination, similarly to several HPV-associated cancers. Many HPV-associated cancers, such as cancers of the cervix, head and neck, and anogenital tract, are accessible to intratumoral injection compared to tumors buried deeper in the body such as pancreatic cancer or lung cancer. Therefore, our finding that intratumoral vaccine administration may be more effective than intramuscular administration creates the distinct clinical opportunity to treat HPV-associated cancers via intratumoral injection. Because TA-CIN has been shown to be safe through intramuscular injection [12], the intratumoral injection of TA-CIN in these HPV-associated malignancies is also likely to be safe. In the current studies, we also demonstrated that intratumoral TA-CIN vaccination can generate systemic antitumor immunity to control distant tumor. Taken together, our results indicate that intratumoral TA-CIN vaccination may be an attractive therapeutic approach for HPV-associated diseases and malignancies.
In the current studies, we have observed that intratumoral injection of TA-CIN was able to generate potent antitumor effects, resulting in a better survival of treated mice (see Fig. 5). The impressive therapeutic antitumor effects are unlikely due to the needle trauma alone. It is conceivable that the damage to the tumor caused by the insertion and removal of the syringe needle may result in damage associated signals, contributing to the anti-tumor immunological response. However, findings by other groups with other antigenic system suggest that immunological responses to intratumoral vaccination is dependent on the vaccine rather than the needle trauma alone, as intratumorally injection of placebos failed to generate the therapeutic antitumor effects comparable to their therapeutic vaccine when injected intratumorally [30,31,32]. Thus, it is unlikely that the immunological enhancements and antitumor effects gained from intratumoral injection of TA-CIN are due to needle damage alone.
In the current study, our data indicate that cross presentation of the HPV antigen is necessary to observe the antitumor effects of the TA-CIN vaccine (Fig. 5). As the major antigen-presenting cells for the induction of T cell adaptive responses, DCs induce cancer immunity by presenting tumor antigens to major histocompatibility complex classes I and II and by expressing costimulatory molecules [26]. Among all kinds of DCs, Batf3-dependent conventional DC type 1 (cDC1) cross-presents tumor-associated antigens exceptionally well [26]. Several studies have shown that Batf3-dependent DCs are required for different cancer immunotherapies, such as checkpoint inhibitors and adoptive T cell transfer therapy [33, 34]. We therefore hypothesized that the antitumor immunity induced by intratumoral TA-CIN vaccination is dependent on Batf3 signaling of cDC1s. Indeed, Batf3 knockout mice completely reversed tumor-specific CD8 + T cell response and antitumor effects of intratumoral TA-CIN vaccination.
We also demonstrated that intratumoral TA-CIN vaccination induces powerful antigen-specific CD8 + T cell responses. To further enhance its therapeutic potency, we sought to combine it with other immunotherapeutic strategies. This is not a novel approach; others have reported increased anti-tumor immune responses when a therapeutic HPV vaccine is intramuscularly injected in combination with PD-L1 checkpoint blockade [35]. Immune checkpoint blockade is a great candidate for combination treatments because the inhibitory immune signaling is a common mechanism for tumor cells to fight back against T-cell mediated immunotherapy [27]. Indeed, our results have shown that intratumoral TA-CIN vaccination increases PD-L1 expression by TC-1 tumor cells and PD-1 expression by TC-1 tumor-infiltrating E7-specific CD8 + T cells. The addition of checkpoint blockades enhanced the vaccine’s anti-tumor immunogenicity by reversing the effects of vaccine-initiated PD-L1/PD-1 increase on CD8 + T cells. Our data have shown that the combination elicits much stronger antitumor immunity compared to either anti-PD-1 antibodies or TA-CIN vaccination alone. Given the promising trend of immune checkpoint blockade in cancer treatment, our combination strategy of intratumoral injection of a cancer vaccine with checkpoint inhibitors may be effective for the treatment of cancer patients who are refractory to checkpoint inhibitors alone as a result of low CD8 + T cell infiltration.
In summary, our results show that intratumoral injection of TA-CIN vaccine can induce a strong E7-specific CD8 + T cell response both locally and systemically. When combining the intratumoral TA-CIN vaccine with PD-1 blockade, we show that combination therapy has much stronger antitumor effects compared to either TA-CIN vaccination or anti-PD-1 antibody administration alone. Our results provide a foundation for future clinical testing of intratumoral TA-CIN vaccination in combination with PD-1 blockade for the control of HPV-associated tumors.
Data availability
Data generated from this study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
References
Parkin DM (2006) The global health burden of infection-associated cancers in the year 2002. Int J Cancer 118(12):3030–3044. https://doi.org/10.1002/ijc.21731
Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N (1999) Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 189(1):12–19. https://doi.org/10.1002/(SICI)1096-9896(199909)189:1%3c12::AID-PATH431%3e3.0.CO;2-F
Safaeian M, Solomon D, Castle PE (2007) Cervical cancer prevention–cervical screening: science in evolution. Obstet Gynecol Clin North Am 34(4):739–760. https://doi.org/10.1016/j.ogc.2007.09.004 ((ix))
Schiller JT, Lowy DR (2012) Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat Rev Microbiol 10(10):681–692. https://doi.org/10.1038/nrmicro2872
Hildesheim A, Herrero R, Wacholder S, Rodriguez AC, Solomon D, Bratti MC, Schiller JT, Gonzalez P, Dubin G, Porras C, Jimenez SE, Lowy DR, Costa Rican HPVVTG (2007) Effect of human papillomavirus 16/18 L1 viruslike particle vaccine among young women with preexisting infection: a randomized trial. JAMA 298(7):743–753. https://doi.org/10.1001/jama.298.7.743
Markman M (2013) Chemoradiation in the management of cervix cancer: current status and future directions. Oncology 84(4):246–250. https://doi.org/10.1159/000346804
Roden RBS, Stern PL (2018) Opportunities and challenges for human papillomavirus vaccination in cancer. Nat Rev Cancer 18(4):240–254. https://doi.org/10.1038/nrc.2018.13
van der Burg SH, Kwappenberg KM, O’Neill T, Brandt RM, Melief CJ, Hickling JK, Offringa R (2001) Pre-clinical safety and efficacy of TA-CIN, a recombinant HPV16 L2E6E7 fusion protein vaccine, in homologous and heterologous prime-boost regimens. Vaccine 19(27):3652–3660. https://doi.org/10.1016/s0264-410x(01)00086-x
Yugawa T, Kiyono T (2009) Molecular mechanisms of cervical carcinogenesis by high-risk human papillomaviruses: novel functions of E6 and E7 oncoproteins. Rev Med Virol 19(2):97–113. https://doi.org/10.1002/rmv.605
Hitzeroth II, Passmore JA, Shephard E, Stewart D, Muller M, Williamson AL, Rybicki EP, Kast WM (2009) Immunogenicity of an HPV-16 L2 DNA vaccine. Vaccine 27(46):6432–6434. https://doi.org/10.1016/j.vaccine.2009.06.015
Wang JW, Hung CF, Huh WK, Trimble CL, Roden RB (2015) Immunoprevention of human papillomavirus-associated malignancies. Cancer Prev Res (Phila) 8(2):95–104. https://doi.org/10.1158/1940-6207.CAPR-14-0311
de Jong A, O’Neill T, Khan AY, Kwappenberg KM, Chisholm SE, Whittle NR, Dobson JA, Jack LC, St Clair Roberts JA, Offringa R, van der Burg SH, Hickling JK (2002) Enhancement of human papillomavirus (HPV) type 16 E6 and E7-specific T-cell immunity in healthy volunteers through vaccination with TA-CIN, an HPV16 L2E7E6 fusion protein vaccine. Vaccine 20(29–30):3456–3464. https://doi.org/10.1016/s0264-410x(02)00350-x
Daayana S, Elkord E, Winters U, Pawlita M, Roden R, Stern PL, Kitchener HC (2010) Phase II trial of imiquimod and HPV therapeutic vaccination in patients with vulval intraepithelial neoplasia. Br J Cancer 102(7):1129–1136. https://doi.org/10.1038/sj.bjc.6605611
Davidson EJ, Faulkner RL, Sehr P, Pawlita M, Smyth LJ, Burt DJ, Tomlinson AE, Hickling J, Kitchener HC, Stern PL (2004) Effect of TA-CIN (HPV 16 L2E6E7) booster immunisation in vulval intraepithelial neoplasia patients previously vaccinated with TA-HPV (vaccinia virus encoding HPV 16/18 E6E7). Vaccine 22(21–22):2722–2729. https://doi.org/10.1016/j.vaccine.2004.01.049
Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU, Haymaker C, Bernatchez C, Curran M, Zecchini Barrese T, Rodriguez Canales J, Wistuba I, Li L, Wang J, van der Burg SH, Melief CJ, Glisson B (2019) Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol 5(1):67–73. https://doi.org/10.1001/jamaoncol.2018.4051
Mkrtichyan M, Chong N, Abu Eid R, Wallecha A, Singh R, Rothman J, Khleif SN (2013) Anti-PD-1 antibody significantly increases therapeutic efficacy of Listeria monocytogenes (Lm)-LLO immunotherapy. J Immunother Cancer 1:15. https://doi.org/10.1186/2051-1426-1-15
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264. https://doi.org/10.1038/nrc3239
Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, Fu YX (2016) Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 29(3):285–296. https://doi.org/10.1016/j.ccell.2016.02.004
Steele KE, Tan TH, Korn R, Dacosta K, Brown C, Kuziora M, Zimmermann J, Laffin B, Widmaier M, Rognoni L, Cardenes R, Schneider K, Boutrin A, Martin P, Zha J, Wiestler T (2018) Measuring multiple parameters of CD8+ tumor-infiltrating lymphocytes in human cancers by image analysis. J Immunother Cancer 6(1):20. https://doi.org/10.1186/s40425-018-0326-x
FDA approves pembrolizumab for advanced cervical cancer with disease progression during or after chemotherapy. (2018) fda.gov. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-advanced-cervical-cancer-disease-progression-during-or-after-chemotherapy. 2020
Hammerich L, Bhardwaj N, Kohrt HE, Brody JD (2016) In situ vaccination for the treatment of cancer. Immunotherapy 8(3):315–330. https://doi.org/10.2217/imt.15.120
Li H, Yu J, Wu Y, Shao B, Wei X (2020) In situ antitumor vaccination: targeting the tumor microenvironment. J Cell Physiol. https://doi.org/10.1002/jcp.29551
Nobuoka D, Yoshikawa T, Takahashi M, Iwama T, Horie K, Shimomura M, Suzuki S, Sakemura N, Nakatsugawa M, Sadamori H, Yagi T, Fujiwara T, Nakatsura T (2013) Intratumoral peptide injection enhances tumor cell antigenicity recognized by cytotoxic T lymphocytes: a potential option for improvement in antigen-specific cancer immunotherapy. Cancer Immunol Immunother 62(4):639–652. https://doi.org/10.1007/s00262-012-1366-6
Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC (1996) Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Can Res 56(1):21–26
Marabelle A, Kohrt H, Caux C, Levy R (2014) Intratumoral immunization: a new paradigm for cancer therapy. Clin Cancer Res 20(7):1747–1756. https://doi.org/10.1158/1078-0432.CCR-13-2116
Sanchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Perez-Gracia JL, Sanchez-Arraez A, Sancho D, Melero I (2017) Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol 28(suppl_12):xii44–xii55. https://doi.org/10.1093/annonc/mdx237
Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK (2017) PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol 8:561. https://doi.org/10.3389/fphar.2017.00561
Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW Jr, Kim Y (2015) STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med 7(283):283ra252. https://doi.org/10.1126/scitranslmed.aaa4306
Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ (2014) Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Can Res 74(15):4042–4052. https://doi.org/10.1158/0008-5472.Can-13-2685
Abdel-Motal UM, Wigglesworth K, Galili U (2009) Intratumoral injection of alpha-gal glycolipids induces a protective anti-tumor T cell response which overcomes Treg activity. Cancer Immunol Immunother 58(10):1545–1556. https://doi.org/10.1007/s00262-009-0662-2
Ishida E, Lee J, Campbell JS, Chakravarty PD, Katori Y, Ogawa T, Johnson L, Mukhopadhyay A, Faquin WC, Lin DT, Wirth LJ, Pierce RH, Pai SI (2019) Intratumoral delivery of an HPV vaccine elicits a broad anti-tumor immune response that translates into a potent anti-tumor effect in a preclinical murine HPV model. Cancer Immunol Immunother 68(8):1273–1286. https://doi.org/10.1007/s00262-019-02357-1
Newman JH, Chesson CB, Herzog NL, Bommareddy PK, Aspromonte SM, Pepe R, Estupinian R, Aboelatta MM, Buddhadev S, Tarabichi S, Lee M, Li S, Medina DJ, Giurini EF, Gupta KH, Guevara-Aleman G, Rossi M, Nowicki C, Abed A, Goldufsky JW, Broucek JR, Redondo RE, Rotter D, Jhawar SR, Wang SJ, Kohlhapp FJ, Kaufman HL, Thomas PG, Gupta V, Kuzel TM, Reiser J, Paras J, Kane MP, Singer EA, Malhotra J, Denzin LK, Sant’Angelo DB, Rabson AB, Lee LY, Lasfar A, Langenfeld J, Schenkel JM, Fidler MJ, Ruiz ES, Marzo AL, Rudra JS, Silk AW, Zloza A (2020) Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc Natl Acad Sci USA 117(2):1119–1128. https://doi.org/10.1073/pnas.1904022116
Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, Sancho D, Melero I (2016) Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov 6(1):71–79. https://doi.org/10.1158/2159-8290.CD-15-0510
Spranger S, Dai D, Horton B, Gajewski TF (2017) Tumor-residing BATF3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31(5):711–723. https://doi.org/10.1016/j.ccell.2017.04.003 ((e714))
Chandra J, Dutton JL, Li B, Woo WP, Xu Y, Tolley LK, Yong M, Wells JW, Leggatt GR, Finlayson N, Frazer IH (2017) DNA vaccine encoding HPV16 oncogenes E6 and E7 induces potent cell-mediated and humoral immunity which protects in tumor challenge and drives E7-expressing skin graft rejection. J Immunother (Hagerstown, Md: 1997) 40(2):62–70. https://doi.org/10.1097/cji.0000000000000156
Funding
This work was supported by the National Institutes of Health under award numbers R01CA237067 and P50CA098252.
Author information
Authors and Affiliations
Contributions
SP contributed to the design and conduction of the experiment. MT contributed to the interpretation of the data. Y-DL contributed to the performance of the experiments. MAC and EF contributed to the original draft preparation. LF, SG, and MT contributed to substantial review and preparation of the manuscript. RBSR contributed to the design of the study. C-FH contributed to the design of the study and interpretation of data. T-CW supervised the study.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
All procedures were performed under a prior-approved protocol of the Johns Hopkins Animal Care and Use Committee, and in accordance with recommendations for the proper use and care of laboratory animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peng, S., Tan, M., Li, YD. et al. PD-1 blockade synergizes with intratumoral vaccination of a therapeutic HPV protein vaccine and elicits regression of tumor in a preclinical model. Cancer Immunol Immunother 70, 1049–1062 (2021). https://doi.org/10.1007/s00262-020-02754-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-020-02754-x