
Abstract
The interaction between programmed cell death protein 1 (PD-1) on activated T cells and its ligands on a target tumour may limit the capacity of chimeric antigen receptor (CAR) T cells to eradicate solid tumours. PD-1 blockade could potentially enhance CAR T cell function. Here, we show that mesothelin is overexpressed in human triple-negative breast cancer cells and can be targeted by CAR T cells. To overcome the suppressive effect of PD-1 on CAR T cells, we utilized CRISPR/Cas9 ribonucleoprotein-mediated editing to disrupt the programmed cell death-1 (PD-1) gene locus in human primary T cells, resulting in a significantly reduced PD-1hi population. This reduction had little effect on CAR T cell proliferation but strongly augmented CAR T cell cytokine production and cytotoxicity towards PD-L1-expressing cancer cells in vitro. CAR T cells with PD-1 disruption show enhanced tumour control and relapse prevention in vivo when compared with CAR T cells with or without αPD-1 antibody blockade. Our study demonstrates a potential advantage of integrated immune checkpoint blockade with CAR T cells in controlling solid tumours and provides an alternative CAR T cell strategy for adoptive transfer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chimeric antigen receptor (CAR) is an artificial molecule containing an antibody-based extracellular structure and cytosolic domains that encode signal transduction modules of the T cell receptor [1]; the CAR targets T cells in an MHC-unrestricted manner, regardless of the status of co-stimulatory factors. The impressive clinical outcomes associated with the adoptive transfer of CAR-expressing T (CAR T) cells in B-lineage malignancy therapy has prompted its application in the treatment of solid tumours. However, efforts to apply the CD19-targeting CAR therapy for haematopoietic malignancies towards the treatment of solid tumours has encountered difficulties [2]. The development of a suitable T cell population that appropriately recognizes cancer antigens without causing detrimental effects to normal cells remains a major challenge [3].
Mesothelin (Meso) is a glycosylphosphatidylinositol-anchored cell surface protein that is expressed at low levels on normal mesothelial cells. Meso knockout mice exhibit normal development and reproduction [4]. Clinical and preclinical data have shown that Meso is overexpressed in mesothelioma, ovarian cancer, pancreatic cancer, and other cancers. Its aberrant expression not only promotes cancer cell proliferation but also contributes to tumour invasion, metastasis and drug resistance; therefore, Meso has emerged as an attractive target for cancer immunotherapy [5]. Several Meso-targeted immunotherapy clinical trials have shown the safety of this treatment [6]. Unlike breast cancer that expresses steroid receptors or HER-2/neu cancer, which responds to many treatments, triple-negative breast cancer (TNBC, which is oestrogen receptor-, progesterone receptor- and human epidermal growth factor receptor 2-negative breast cancer) is largely unresponsive to the clinically available targeted therapies and frequently has the worst prognosis among breast tumour subtypes [7]. Meso is overexpressed in 34–67% of TNBC patients [8,9,10] but is relatively rare in ER+ or Her2/neu+ breast cancer patients. Meso-positive TNBC patients develop more distant metastases and have a shorter disease-free interval with significantly lower overall and disease-free survival rates than Meso-negative TNBC patients [11]. Tchou et al. have previously shown that Meso CAR T cells display cytotoxic activity towards TNBC cell lines highly expressing Meso [9], but their effector function requires further characterization.
It is well accepted that tumours adapt immune regulatory signalling pathways to evade immune recognition and elimination [12]. One of the mechanisms utilized by tumours is the upregulation of programmed death-ligand 1 (PD-L1), which has been identified as an indicator of poor prognosis in several tumour types, including breast cancers. By engaging programmed death 1 (PD-1) receptors on activated T cells, PD-L1+ tumours direct tumour-specific T cells, including adoptively transferred T cells, towards functional exhaustion. We have shown that PD-1 expression causes a step-wise loss of proliferative capacity, cytokine secretion, and finally the cytotoxic activity of T cells [13]. Even with a co-activation signal from 4-1BB, tumour-infiltrated Meso CAR T cells still show a hypofunctional phenotype and rapidly lose their cytokine secretion and cytotoxic abilities. This loss of function is associated with increased expression of the surface inhibitory receptor PD-1, which has been confirmed by PD-1 blockade in vitro with a PD-L1 neutralizing antibody [14]. We hypothesized that blockade of the effector function of CAR T cells with PD-1 could be enhanced. Anti-PD-L1 and PD-1 antibodies have been used in clinical trials and in the treatment of different cancer types. Despite the success of monoclonal antibody blockade of PD-1 in human cancer treatment, most patients do not have favourable clinical responses [15, 16]. In vivo imaging data have revealed that this result is partially because anti-PD-1 monoclonal antibodies are captured prior to reaching the T cell surface by PD-1 tumour-associated macrophages through their Fc domain, removing their ability to block PD-1 and resulting in suppression of T cell function [17]. Several other strategies have also been applied for PD-1 blockade, including PD1/CD28 converters [18, 19], PD-1 dominant-negative receptors [20] and PD-1 knockout by the CRISPR/Cas9 system [21]. The benefit of combining PD-1 blockade treatment with CAR T cells has been observed in several reports [19]. Administration of PD-1 blocking antibody to patients with refractory diffuse large B cell lymphoma and progressive lymphoma has been effective in preventing the failure of CD19-directed CAR T cell therapy [22]. The combination of PD-1 disruption through CRISPR/Cas9 and CD19-CAR T cell therapy against B cell lymphoma and other models has been studied [23,24,25], but whether PD-1 disruption would improve CAR T cell therapy in a breast cancer model has not been fully reported in the literature.
With the goal of overcoming the tumour PD-L1 effects on adoptively transferred T cells, we sought to evaluate T cell effector functions by PD-1 knockdown mediated by a CRISPR/Cas9 approach. Here, we show that CAR T cells that specifically recognize Meso can target Meso-expressing tumours (TNBC) and that this effect is further enhanced by combination with CRISPR/Cas9-mediated PD-1 genome modification.
Materials and methods
T cell isolation, gene transfer, and expansion
Peripheral blood mononuclear cells (PBMCs) were purified by Ficoll-Paque PLUS (GE Healthcare) density centrifugation. The CD4 or CD8 T cells were isolated and expanded as previously described by Wei et al. [13].
Plasmids
Sequences of the Meso-specific target scFv with a CD8 leading sequence, a CD8 hinge and transmembrane sequence, and 4-1BB and CD3ζ signalling domains were synthesized (Thermo Fisher Scientific) and inserted into a lentiviral vector. A DNA fragment containing a PD-1 genomic exon 1 targeting sequence was synthesized, ligated with a manufacturer-provided sgRNA oligonucleotide template and then inserted into the pMDTM 19 T-vector.
Antibody staining and flow cytometry
Surface and intracellular cytokine staining was performed as described above. Data were obtained with an Accuri™ C6 flow cytometer (BD Biosciences) and analysed with FlowJo software (Tree Star Inc.). The following antibodies were used for cell surface and intracellular staining: APC-PD1 (eBioscience, clone J105), FITC-Meso (R&D, clone 420411), FITC-CD4 (BD Biosciences, clone RPA-T4), PE-CD25 (BD Biosciences, clone 4E3), PerCP-CD8 (BD Biosciences, clone RPA-T8), PE-IL-2 (BD Biosciences, clone MQ1-17H12), APC-IFNγ (BD Biosciences, clone B27), CD3 (BD Biosciences, clone UCH71), and PE-CD107a (BD Biosciences, clone H4A3) antibodies. To detect CAR expression, cells were stained using goat anti-mouse IgG-biotin (Jackson ImmunoResearch) followed by streptavidin-PE (BD Biosciences).
Cytotoxicity assays
The cytotoxic capacity of CAR T cells was tested in an 18-h luciferase-based killing assay as described elsewhere [26]. Lysis efficiency was calculated as [1 − (RLUsample)/(RLUmax)] × 100.
PD-1 sgRNA-Cas9 RNP assembly and electroporation
PD-1 sgRNA was transcribed in vitro under the control of a T7 promoter, further purified, and then assembled with GeneArt™ Platinum™ Cas9 Nuclease (Thermo Fisher Scientific). At day 5 after activation, PD-1 sgRNA was electroporated into T cells with a Neon transfection kit and device (Thermo Fisher Scientific).
Analysis of genome editing and PD-1 gene expression
Editing efficiency was estimated by PD-1 surface staining and a T7 endonuclease I assay. At day 14, T cells were stimulated by Dynabeads® Human T-Activator CD3/CD28 (Thermo Fisher Scientific) and cultured in a 37 °C incubator for 48 h. Then, PD-1 and CD25 staining was performed. Data were analysed by flow cytometry. A T7E1 assay was performed following the manufacturer’s instructions. The CRISPR/Cas9 editing efficiency was calculated by the following equation: % gene modification = 100 × (1 − (1 − fraction cleaved)1/2).
Xenograft assays
A total of 2 × 106 firefly luciferase-expressing BT549 cells were injected into the fourth mammary gland of NOD-Prkdcscid IL2rgnull (NSG) mice. Mice were monitored weekly for tumour growth by bioluminescence imaging with a Xenogen Spectrum system and Living Image software version 3.2 (Calipere PerkinElmer, Hopkinton, MA, USA) following a previously described protocol [27]. At day 25, T cells were administered at a dose of 1 × 105 Meso CAR+ T cells/mouse through the tail vein. PD-1-blocking antibody (clone EH12, Biolegend) was injected at 2 mg/kg every 2 weeks through the tail vein. Peripheral blood was drawn from the tail vein and stained with anti-human CD45 antibody (clone H130, Biolegend) at the indicated experimental time points and mixed with CountBright™ Absolute Counting Beads (Thermo Fisher Scientific) as an event collecting control. The samples were analysed by flow cytometry, and cells were quantified by the number of hCD45+ cell events/the number of bead events × number of beads per test/test volume.
Results
Mesothelin and PD-L1 are co-expressed in BT-549 and HeLa cell lines
To study whether gene editing at the pdcd-1 locus would influence the expression of PD-1 and enhance CAR function in T cells, we evaluated Meso and PD-L1 expression levels in MCF7 and three available TNBC lines (MDA-MB-231, BT-549 and Hs 578T). Using HeLa cells as a reference (cell line from which Meso as an antigen was cloned and with which monoclonal antibodies are screened to construct the CAR molecule) [28, 29], Meso was detected in BT-549 cells but not in MCF7, MDA-MB-231 and Hs 578T cells by both Western blot (Fig. 1a) and flow cytometry (Fig. 1b). This finding is consistent with that of previous reports, suggesting that Meso is not expressed in MDA-MB-231 cells [9]. The flow cytometry data (Fig. 1b) showed that all cell lines expressed PD-L1 at various levels. Therefore, we chose Meso+PD-L1+ BT-549 and HeLa cells as putative target cells and Meso−PD-L1+ MCF7 cells as non-target cells to evaluate Meso-specific CAR T cell function.

Evaluation of mesothelin and PD-L1 expression in various cancer cell lines. a Mesothelin expression levels were evaluated by Western blot. b Histograms depicting mesothelin and PD-L1 expression (red line) in the breast cancer oestrogen and progesterone receptor-positive line MCF7 and the TNBC lines MDA-MB-231, BT-549 and Hs 578 T. HeLa cells served as a positive control for mesothelin. Isotype staining served as a negative control for mesothelin and PD-L1 staining (solid grey with dotted line)
Characterization of the effector function of Meso CAR T cells
Carpenito et al. constructed a Meso-targeted CAR containing different signalling domains [30]. CAR T cells with a 4-1BB signalling domain showed the greatest persistence in a tumour model [31] and advanced toward towards a central memory phenotype [32]. We generated a Meso CAR molecule with a Meso-binding scFv domain, 4-1BB intracellular and TCR-ζ signal transduction domains and other required regions in a lentiviral vector (Supplementary Figure 1a) and confirmed Meso CAR expression in human primary T cells (Supplementary Figure 1b).
To investigate the antitumour effects of the Meso CAR T cells, cytotoxicity was measured in both an indirect way by surface CD107a staining (Supplementary Figure 2a) [33] and a direct way by a luciferase-based cytotoxic T lymphocyte (CTL) assay [26]. We transduced MCF7, BT-549 and HeLa cells with a lentivirus containing DNA encoding both red fluorescent protein (RFP) and luciferase. Cells were sorted based on RFP expression, and a cell line stably expressing luciferase was generated as a target for luciferase-based CTL assays (Fig. 2a). At day 9 after stimulation, Meso CAR T cells were mixed with target cells at various effector:target (E:T) ratios and co-incubated for 18 h (Fig. 2b). We observed that 40% of BT-549 cells and 58% of HeLa cells were lysed even at a low E:T ratio of 0.5:1 and that the cytotoxic activity of CAR T cells increased with increasing E:T ratios.

Meso CAR T cell effects on mesothelin-expressing tumour cells. a Luciferase-RFP stably transduced cell lines (MCF7, BT-549, and HeLa) were generated (red line). The untransduced parent cell lines are depicted with black lines. Lower panel: b flow cytometry-based cytotoxicity assay. Control or Meso CAR T cells were co-incubated with different targets for 18 h at the indicated E:T ratios. Primary human T cells were transduced with lentivirus encoding Meso CAR. Nine days later, T cells were co-cultured with the indicated cells. The cytotoxicity of T cells was measured by luciferase assay. c Levels of interferon-γ (IFN-γ) and d interleukin-2 (IL-2) were measured by enzyme-linked immunosorbent assay (ELISA) 24 h after culturing control or Meso CAR T cells with MCF7, BT-549 and HeLa at E:T ratios of 1:1 and 1:2. Data are reported as the mean from three independent experiments. Error bars indicate the SEM
The antitumour effects of CAR-redirected T cells depend on their capacity to secrete cytokines after exposure to antigens. Therefore, cytokine secretion from Meso CAR T cells was measured by intracellular cytokine staining (Supplementary Figure 2b) and enzyme-linked immunosorbent assay (ELISA) after exposure to target cells. We detected robust IFNγ (Fig. 2c) and IL-2 (Fig. 2d) secretion when CAR T cells were co-incubated with BT549 or HeLa cells but not with MCF7 cells.
These results show that Meso CAR T cells had potent effects on Meso-expressing TNBC cells.
Ablation of the T cell negative regulator PD-1 with Cas9 ribonucleoproteins (RNPs)
Expression of the exhaustion marker/inhibitory receptor PD-1 has been observed in 19BBz-CAR T cells, even though 4-1BB pathways showed a reduced induction of PD-1 expression [31]. Due to the upregulation of PD-1 expression on the surface, tumour-infiltrated CAR T cells could rapidly lose their antitumour cell function [14]. To simultaneously express CAR and knockout pdcd1, we first stimulated and transduced human T cells (CD4 mixed with CD8 at a ratio of 1:1) as described previously [13] and subsequently electroporated PD-1 sgRNA/Cas9 RNP assembly 5 days later when the pdcd1 locus was still open for PD-1 expression (a protocol for combined lentiviral and Cas9 RNP transduction of human primary T cells is schematically shown in Fig. 3a, and the targeting locus is indicated in Fig. 3b). IL-7 (5 ng/ml) and low concentrations of IL-2 (10 U/ml) were added to the culture medium to yield a better cell survival rate after electroporation. The T cells were re-stimulated by Dynabeads® Human T-Activator CD3/CD28 at day 12 and stained with anti-PD-1 antibody 2 days later. PD-1 expression was analysed within the activated cell population (gated with CD25+). A 59.2 ± 9.0% (n = 6) reduction in the PD-1hi population was observed in Meso CAR/PD-1 sgRNA-Cas9 RNP cells when compared with Meso CAR T cells (Fig. 3c). A 30.8 ± 5.0% (n = 5) disruption was also confirmed with the T7E1 assay (which might underestimate knockout efficiency) (Fig. 3d) [21]. There was no difference in PD-1 disruption efficiency between the Meso CAR+ and Meso CAR− populations (Supplementary Figure 3). To determine whether PD-1 disruption would impact T cell persistence, we focused on the fraction of CD8 T cells after in vitro expansion (Fig. 3e). A bias towards CD8 T cells was noted in control cells, since IL-2 was added to the culture medium to promote CD8 T cell growth. Interestingly, the addition of IL-2 did not increase the fraction of CD8 T cells in Meso CAR-expressing T cells as significantly as in the control cells. Even with the 4-1BB signal, which supports a moderate rise in the CD8 T cell fraction, the Meso CAR T cell groups (with or without PD-1 sgRNA-Cas9 RNP) showed lower CD8 counts than the control group, which suggests that the electroporation procedure might affect the growth of CD8 T cells more than that of CD4 T cells. The Meso CAR/PD-1 sgRNA-Cas9 RNP T cells showed the lowest CD8 fraction, which indicated that PD-1 knockout might affect CD8 T cell expansion for unknown reasons. No difference was observed in the transduction efficiency of Meso CAR in individual CD4 or CD8 subsets between Meso CAR/PD-1 sgRNA-Cas9 RNP and Meso CAR only groups (data not shown).

PD-1 gene editing by sgRNA/Cas9 RNP assembly in primary human T cells. a Schematic representation of the experimental protocol of introducing PD-1 sgRNA/Cas9 RNP assembly and CAR into T cells. b Schematic representation of the PD-1 sgRNA/Cas9 RNP assembly-targeted site in the human PDCD-1 genome. c PD-1 expression is shown by histograms of PD-1 cell-surface staining. The PD-1 sgRNA/Cas9 RNP assembly was electroporated into T cells 5 days after stimulation, and PD-1 expression was then evaluated on day 2 after the second stimulation. The mean fluorescence intensity of PD-1 staining is shown above each sample. (*p < 0.05, **p < 0.01, Student’s t test). d The T7E1 assay was used to confirm successful editing in the PD-1 genome locus. e PD-1 editing significantly affects the CD8 T cell fraction (*p < 0.05, **p < 0.01, Student’s t test). CD8 T cells were analysed by flow cytometry
PD-1 disruption in CAR T cells enhances their ex vivo effector functions
To determine whether PD-1 disruption influences T cell activity, CAR T cells with/without PD-1 disruption were washed extensively with phosphate-buffered saline to remove the residual IL-2 at day 14 and then co-incubated with MCF7, BT-549 or HeLa cells at a ratio of 1:1 unless indicated otherwise. When BT-549 and HeLa cells were co-cultured with T cells expressing Meso CAR for 18 h, they showed 11.44- and 3.8-fold increases in PD-L1 expression, respectively (Fig. 4a). Expression of PD-L1 in tumour cells could be further stimulated (without significant differences) when the tumour cells were co-cultured with Meso CAR/PD-1 sgRNA-Cas9 RNP T cells. No elevation in PD-L1 expression was observed in MCF7 cells. We next examined the cytokine production and cytotoxic capacities of Meso CAR/PD-1 sgRNA-Cas9 RNP T cells. When exposed to antigens expressed on BT-549 and HeLa cells at 2 different ratios, Meso CAR/PD-1 sgRNA-Cas9 RNP T cells showed significantly higher antitumour activity (Fig. 4b) than Meso CAR T cells; however, both groups showed a reduction in cytotoxicity due to the extended cell culture time. The actual enhancement of cytotoxic capacity per cell could be greater considering the lower CD4/CD8 ratio shown in Fig. 3e. Meso CAR/PD-1 sgRNA-Cas9 RNP T cells showed significantly elevated IFN-γ and IL-2 levels (Fig. 4c) compared with control CAR T cells. The elevated IFN-γ levels were also reflected by the increased expression of PD-L1 in tumour cells, since IFN-γ could stimulate PD-L1 expression. We did not observe improved in vitro expansion from PD-1 disruption when Meso CAR/PD-1 sgRNA-Cas9 RNP T cells were stimulated by BT-549 at day 14 and day 19 (indicated as the 1st and 2nd time points) after the first expansion following the application of Dynabeads® Human T-Activator CD3/CD28 (Fig. 4d), which suggested that PD-1 disruption may have differential effects on different T cell effector functions, which may be correlated with our previous finding [13]. There was no obvious alteration in the levels of other co-inhibitory factors, including CTLA-4 and Tim-3 (Fig. 4e), which suggested that the enhanced effects were attributed to only PD-1 Cas9 RNP-induced PD-1 disruption.

Enhanced Meso CAR T cell effect upon PD-1 gene disruption. a Increased expression of PD-L1 in the cancer cell lines MCF7, BT-549 and HeLa was observed when cells were co-cultured separately with Meso CAR or Meso CAR/PD-1 sgRNA/Cas9 RNP T cells for 18 h. (*p < 0.05, **p < 0.01, Student’s t test). b The cytotoxicity of T cells was measured by luciferase assay. c After co-culture for an additional 30 h, the supernatant was collected, and the ability of these stimulated T cells to produce IFNγ and IL-2 was measured by enzyme-linked immunosorbent assay. b, c Data (mean ± SEM) are representative of at least 3 independent experiments. Statistically significant effector function enhancement in PD-1-edited CAR T cells was observed at 2 different E:T ratios (*p < 0.05, **p < 0.01, Student’s t test). d At day 14, T cells were stimulated by BT-549 cells (first stimulation). Five days later, the cells were re-stimulated by BT-549 cells (second stimulation). The ratio of BT-549:T cells was 1:3, and surviving T cells were counted after trypan blue staining. Data are representative of at least three independent experiments with different donors and plotted as the mean ± SEM. e CTLA-4 and Tim-3 expression in T cells was compared between Meso CAR (dark line) and Meso CAR/PD-1 sgRNA/Cas9 RNP T cells (red lines)
As PD-1 blocking antibodies have demonstrated beneficial effects in the treatment of various malignancies, including several solid tumours, we next compared the abilities of PD-1 editing by Cas9 RNP and PD-1 blocking antibody (clone EH12.2H7) to rescue CAR T cell function (Fig. 5). PD-1 disruption by sgRNA-Cas9 RNP exhibited significantly higher potency in enhancing cytotoxicity and the production of cytokines, IL-2 and IFNγ by Meso CAR T cells when compared that by antibody blockade.

PD-1-edited CAR T cells show better cytotoxicity and cytokine production than PD-1 blocking antibody-treated CAR T cells. Meso CAR T cells with and without PD-1 sgRNA/Cas9 RNP or αPD-1 blocking antibody treatment were used in functional assays. The αPD-1 blocking antibody EH12.2H7 was added at 10 µg/ml when T cells were mixed with BT549 cells
PD-1-disrupted CAR T cells exhibit better tumour control in vivo
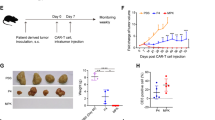
Next, we evaluated whether Meso CAR/PD-1 sgRNA-Cas9 RNP T cells would provide an in vivo advantage in the orthotopic xenograft NOD-Prkdcscid IL2rgnull (NSG) mouse model utilizing BT-549 cells stably expressing luciferase. The treatment response was monitored by tumour burden measurements (bioluminescence imaging, BLI) and T cell persistence (hCD45+ cell counts and CAR genomic copies). A low dose (1 × 105 per mouse) of Meso CAR+ T cells was injected into mice with a pre-established tumour burden. Meso CAR/PD-1 sgRNA-Cas9 RNP T cells showed a better antitumour effect than Meso CAR T cells (Fig. 6a), with a significant decrease in tumour BLI at day 18 (Fig. 6b). An increasing hCD45+ cell count was observed during the clearance of tumour cells, but the difference was not significant between groups (Fig. 6c); however, there was a significant increase in the number of Meso CAR genomic copies at day 18 (Fig. 6d), which suggested better expansion of Meso CAR T cells in vivo with PD-1 editing. To compare the effect of the PD-1 blocking antibody vs. that of PD-1 editing on rescuing CAR T cells, EH12 antibody was subsequently administered intraperitoneally at a concentration of 2 mg/kg to the indicated group starting at day 1 every 2 weeks following the clinical protocol described previously [22]. The results showed that PD-1 blocking antibody did not improve the effect of CAR T cells in our model (Fig. 6a, b). Tumour relapse was observed in all groups at day 44 and was the most severe in the Meso CAR/αPD-1 group and the least severe in the Meso CAR/PD-1 sgRNA-Cas9 RNP group. To clarify whether the relapse was caused by antigen loss in the tumour cells or a lack of T cell persistence, we sacrificed mice and dissected the tumour (Fig. 6e) to analyse hCD3+ T cell infiltration (Fig. 6f) and the expression of Meso in the tumour (Fig. 6g). We observed significantly greater hCD3+ T cell infiltration in the tumour of Meso CAR/PD-1 sgRNA-Cas9 RNP T cell group than in that of the Meso CAR T cell group. A decrease in Meso expression was observed in the majority of samples, which indicated that the relapse might have resulted from the loss of antigen. However, we also observed increased expression of Meso in two out of four samples in the Meso CAR T cell/αPD-1 group, along with the presence of CD3 T cells in the tumour and Meso CAR copies in the peripheral blood. These observations suggest that T cell exhaustion may have occurred in the Meso CAR T cell/αPD-1 group. By the end of the experiment, we did not observe any autoimmune symptoms among all treated groups.

PD-1 editing shows better synergy with the CAR in T cell-mediated tumour control in vivo when compared with PD-1 blocking antibody treatment. a Enhanced CAR T cell tumour control capacity with sgRNA/Cas9 RNP-mediated PD-1 disruption in NSG mice with a BT549 orthotopic xenograft model. b The enhanced efficacy was measured by BLI in each group of mice. c Human CD45+ cells in peripheral blood collected from the tail vein were separated by flow cytometry and quantified by CountBright™ Absolute Counting Beads. The results are expressed as the mean count ± SEM with n = 8 for each group. d Meso CAR copies were measured in genomic DNA extracted from mouse blood samples at the indicated time points by absolute quantitative PCR (*p < 0.05, **p < 0.01, Student’s t test). e Tumours were dissected from mice at day 44 after CAR T cell treatment. f hCD3+ cells were quantified from single-cell suspensions from collagenase VIII- and DNase-digested tumour samples (*p < 0.05, **p < 0.01, Student’s t test). g Expression of mesothelin in dissected tumours from each group was measured by Western blot
Thus, PD-1 disruption through CRISPR Cas9 enhanced the capacity of CAR T cells to control large established tumours.
Discussion
Although two major cancer immunotherapy approaches, CAR-engineered T cells and blockade of co-inhibitory factors, have provided cancer patients with more hope for curative therapies, control of disease progression in many cancer types, especially solid tumours, remains a great challenge. CAR T cells have been reported as hypofunctional in the tumour microenvironment [14] partially due to the suppressive effect of their own expression of PD-1. The combination of CAR T cells and PD-1 blockade could improve tumour control and overall survival [34]. PD-1 blockade achieved by blocking antibodies, dominant-negative receptors, or chimeric PD-1 switching molecules may improve CAR T cell efficacy. The blockade efficacy of PD-1 blocking antibody is short-lived and relies upon repeated administration [15, 20]. Recently, reports have shown that CRISPR/Cas9-mediated disruption of PD-1 could boost the function of CD19-CAR T cells [24, 35] and that Epstein–Barr virus LMP2 peptide increased the cytotoxic function of T lymphocytes to different degrees [25]. In the present study, we chose to introduce PD-1 sgRNA/Cas9 RNP cells after a single stimulation with αCD3/CD28 beads, which resulted in shorter in vitro culture time and preservation of Cas9 editing efficiency when compared with two longer stimulations with αCD3/CD28 beads using the protocol provided by Rupp et al. [24]. We also showed that Meso CAR T cells could be genetically edited at the PD-1 locus, enhancing their capacities for cytotoxicity and cytokine secretion against TNBC. In particular, we observed a 2- to threefold increase in cytotoxicity to BT549 cells, which is a much greater enhancement compared with that in a previous report [31]. However, this gene editing protocol did not alter the proliferative ability of the cells or the expression of the co-inhibitory factors CTLA-4 and Tim-3.
It has been shown that different Meso antibody clones can display variable reactivity on the cell membrane and in the cytoplasm of neoplastic cells in the same samples [36]. The inconsistency between the poor surface protein staining by flow cytometry and strong Meso protein staining by Western blot suggests that our antibody may not efficiently bind the cell surface or that there is less surface expression of Meso. Nevertheless, a dramatic difference in the responses of Meso CAR T cells to different Meso expression levels in target cells was clearly observed, both in terms of cytotoxicity and cytokine production. Interestingly, June’s group observed that CTL019 (CD19-CAR T) cells were cytotoxic to multiple myeloma neoplastic cells and could be successfully utilized in the treatment of one patient with refractory multiple myeloma [37]; however, the patient’s neoplastic plasma cells expressed extremely low levels of CD19. This finding suggests that the efficacy of immunotherapy treatments in targeting shared antigens, especially in the heterogeneous solid tumour background, may need additional evaluation.
Upon PD-1 disruption, Meso CAR T cells showed increased cytotoxicity and cytokine production but not increased proliferation in vitro. This observation could be related to the low expression levels of Meso at the surface of the tumour cells. This observation could also be correlated with our previous findings [13]. The capacity of T cell proliferation was more suppressed than other T cell functions by PD-1-mediated signals when PD-L1-expressing artificial antigen-presenting cells (K562) were used to stimulate resting SL9 TCR-expressing T cells with barely detectable levels of endogenous PD-1 [13]. We observed no alterations in the expression of the co-inhibitory factors CTLA-4 and Tim-3. In a lymphocytic choriomeningitis virus (LCMV) chronic infection and tumour model, exhausted T cells showed remarkably stable exhaustion-associated DNA methylation programming, which restricted T cell clonal expansion in response to PD-1 blockade therapy [38]. These data suggested that alterations in the PD-1/PD-L1 axis did not influence the DNA methylation status across the genomic region related to proliferation. Therefore, T cell PD-1 disruption using our approach may not have improved T cell proliferation via changes to DNA methylation status, which needs further elucidation.
The phenomenon of relapse after CAR T cell treatment has been observed in various clinical reports. Our data suggested that PD-1 disruption through CRISPR/Cas9 could not only enhance tumour control by Meso CAR T cells but also delay tumour relapse to some degree after CAR T cell treatment. Our study provides evidence that the combination of CAR T cells and co-inhibitory factor gene editing is effective.
We showed that PD-1 disruption mediated by CRISPR/Cas9 improved the effect of the CAR, which contains 4-1BB and CD3 (BBz) as signalling domains, on T cell function. Although Meso 28z and BBz-CAR T cells exhibited equivalent secretion of effector cytokines and proliferation in vitro upon initial antigen stimulation, Meso 28z-CAR T cells expressed higher levels of exhaustion markers, such as PD-1, TIM-3, and LAG-3, upon further stimulation than Meso BBz-CAR T cells [20]. We anticipate that PD-1 disruption could further enhance the function of CAR T cells containing the CD28 and CD3 signalling domain (28z).
Overall, we have demonstrated that PD-1 disruption mediated by Cas9/CRISPR could improve CAR T cell effector function in vitro and in vivo. A combination of positive stimulation from the CAR and blockade of negative regulators, such as PD-1, could enhance T cell antitumour effects against TNBC.
Abbreviations
- 4-1BB TNF:
-
Receptor superfamily member 9
- CRISPR/Cas9:
-
sgRNA-guided clustered regularly interspaced short palindrome repeats-associated nuclease Cas9
- Meso:
-
Mesothelin
- RFP:
-
Red fluorescence protein
- RNP:
-
Ribonucleoprotein
- T7E1:
-
T7 endonuclease I
- Tim-3:
-
T cell immunoglobulin and mucin-domain-containing-3
- TNBC:
-
Triple-negative breast cancer
References
Gross G, Waks T, Eshhar Z (1989) Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 86(24):10024–10028
Johnson LA, June CH (2017) Driving gene-engineered T cell immunotherapy of cancer. Cell Res 27(1):38–58
Gross G, Eshhar Z (2016) Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol 56:59–83
Bera TK, Pastan I (2000) Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol 20(8):2902–2906
Morello A, Sadelain M, Adusumilli PS (2016) Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov 6(2):133–146
Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, Kalos M, June CH (2014) Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2(2):112–120
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig 121(7):2750–2767
Parinyanitikul N, Blumenschein GR, Wu Y, Lei X, Chavez-Macgregor M, Smart M, Gonzalez-Angulo AM (2013) Mesothelin expression and survival outcomes in triple receptor negative breast cancer. Clin Breast Cancer 13(5):378–384
Tchou J, Wang LC, Selven B, Zhang H, Conejo-Garcia J, Borghaei H, Kalos M, Vondeheide RH, Albelda SM, June CH, Zhang PJ (2012) Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat 133(2):799–804
Lamberts LE, de Groot DJ, Bense RD, de Vries EG, Fehrmann RS (2015) Functional genomic mRNA profiling of a large cancer data base demonstrates mesothelin overexpression in a broad range of tumor types. Oncotarget 6(29):28164–28172
Tozbikian G, Brogi E, Kadota K, Catalano J, Akram M, Patil S, Ho AY, Reis-Filho JS, Weigelt B, Norton L, Adusumilli PS, Wen HY (2014) Mesothelin expression in triple negative breast carcinomas correlates significantly with basal-like phenotype, distant metastases and decreased survival. PloS One 9(12):e114900
Motz GT, Coukos G (2013) Deciphering and reversing tumor immune suppression. Immunity 39(1):61–73
Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, Freeman GJ, Krogsgaard M, Riley JL (2013) Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci USA 110(27):E2480–E2489
Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, June CH, Riley JL, Wherry EJ, Albelda SM (2014) Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric Antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 20(16):4262–4273
Callahan MK, Postow MA, Wolchok JD (2016) Targeting T cell co-receptors for cancer therapy. Immunity 44(5):1069–1078
Borch TH, Donia M, Andersen MH, Svane IM (2015) Reorienting the immune system in the treatment of cancer by using anti-PD-1 and anti-PD-L1 antibodies. Drug Discov Today 20(9):1127–1134
Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, Weissleder R, Pittet MJ (2017) In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aal3604
Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, Moon EK (2016) A Chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res 76(6):1578–1590
Chen N, Morello A, Tano Z, Adusumilli PS (2017) CAR T-cell intrinsic PD-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. Oncoimmunology 6(2):e1273302
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS (2016) Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Investig 126(8):3130–3144
Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, Marson A (2015) Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci USA 112(33):10437–10442
Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, June CH, Schuster SJ (2017) PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129(8):1039–1041
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y (2017) Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res 23(9):2255–2266
Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A (2017) CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 7(1):737
Su S, Zou Z, Chen F, Ding N, Du J, Shao J, Li L, Fu Y, Hu B, Yang Y, Sha H, Meng F, Wei J, Huang X, Liu B (2017) CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology 6(1):e1249558
Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, Boesteanu AC, Cogdill AP, Chen T, Fraietta JA, Kloss CC, Posey AD Jr, Engels B, Singh R, Ezell T, Idamakanti N, Ramones MH, Li N, Zhou L, Plesa G, Seykora JT, Okada H, June CH, Brogdon JL, Maus MV (2015) Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 7(275):275ra222
Zhang Q, Wei F, Wang HY, Liu X, Roy D, Xiong QB, Jiang S, Medvec A, Danet-Desnoyers G, Watt C, Tomczak E, Kalos M, Riley JL, Wasik MA (2013) The potent oncogene NPM-ALK mediates malignant transformation of normal human CD4(+) T lymphocytes. Am J Pathol 183(6):1971–1980
Chowdhury PS, Viner JL, Beers R, Pastan I (1998) Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc Natl Acad Sci USA 95(2):669–674
Chang K, Pastan I (1996) Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA 93(1):136–140
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH (2009) Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA 106(9):3360–3365
Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M (2015) Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer cell 28(4):415–428
Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, Patel PR, Guedan S, Scholler J, Keith B, Snyder NW, Blair IA, Milone MC, June CH (2016) Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44(2):380–390
Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, Zhou L, Loew A, Young RM, June CH, Zhao Y (2015) Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 75(17):3596–3607
Sharma P, Allison JP (2015) Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161(2):205–214
Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, Zhao Y (2017) A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 8(10):17002–17011
Inaguma S, Wang Z, Lasota J, Onda M, Czapiewski P, Langfort R, Rys J, Szpor J, Waloszczyk P, Okon K, Biernat W, Ikeda H, Schrump DS, Hassan R, Pastan I, Miettinen M (2017) Comprehensive immunohistochemical study of mesothelin (MSLN) using different monoclonal antibodies 5B2 and MN-1 in 1562 tumors with evaluation of its prognostic value in malignant pleural mesothelioma. Oncotarget 8(16):26744–26754
Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, Zheng Z, Vogl DT, Cohen AD, Weiss BM, Dengel K, Kerr ND, Bagg A, Levine BL, June CH, Stadtmauer EA (2015) Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 373(11):1040–1047
Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG, Youngblood B (2017) De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170(1):142–157 e119
Acknowledgements
We are grateful to the blood donors for their contribution; Zhouluo Ou for providing the breast cancer cell lines used in this study; Ilya Vinnikov for proofreading the manuscript; and Wei Zhang and Huan Wang for their helpful suggestions.
Funding
This work was supported by the National Natural Science Foundation of China (81402542) and the scholarship of Pujiang Talents in Shanghai to Fang Wei (14PJ1405600).
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to the conception and design of this work. WH, ZZ and YJ performed the experiments, analysed the data and wrote the manuscript. KS contributed to blood donor communication and sample collection. GL contributed to the molecular biology experiments. QC and XM proofread the manuscript and gave intellectual suggestions. FW contributed to the conception and design, data analysis and interpretation, and manuscript writing and provided financial support. All authors viewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
For the use of blood samples from healthy donors, written informed consent was obtained in accordance with the of the Shanghai Jiao Tong Human Sample Committee on March 1, 2014. The mouse study was carried out in accordance with the recommendations of East China Normal University Animal Care guidelines from the East China Normal University Animal Care Committee. All experimental protocols were approved on August 1, 2016, by the East China Normal University Animal Care Committee.
Animal source
Six-week-old female NOD-Prkdcscid IL2rgnull (NSG) mice were purchased from Vitalstar, China, and housed in ventilated cages in our pathogen-free facility.
Cell line authentication
The human cell lines MCF7, MDA-MB-231, BT-549, Hs 578T and HeLa were kindly provided by Zouluo Ou (Fudan University, Shanghai, China). The cell lines were authenticated by the Chinese Academy of Sciences Committee Type Culture Collection Cell Bank.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hu, W., Zi, Z., Jin, Y. et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother 68, 365–377 (2019). https://doi.org/10.1007/s00262-018-2281-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-018-2281-2