
Abstract
Although a role of PD-L1 in the suppression of anti-tumor immunity and its value as a predictive biomarker has been suggested by various preclinical and clinical studies, the precise mechanisms how PD-L1 and PD-L2, another ligand of PD-1, regulate anti-tumor immunity in the tumor microenvironment are yet to be fully explored. Here, we address this issue using PD-L1-deficient tumor cells, PD-L1-knockout (KO) mice, anti-PD-L1 monoclonal antibody (mAb), and anti-PD-L2 mAb. Firstly, PD-L1-deficient or competent tumor cells were inoculated into wild-type or PD-L1-KO mice. Results of tumor growth and mouse survival indicated that both tumor- and host-derived PD-L1 are functional to suppress anti-tumor immunity, while the former contributes predominantly than the latter. Experiments using bone marrow (BM) chimeric mice, generated by transferring PD-L1-KO BM cells into wild-type mice or vice versa, further suggested that PD-L1 expressed on BM-derived hematopoietic cells mediates the suppressive effects on anti-tumor immunity. Secondly, anti-PD-L2 mAb treatment demonstrated a profound synergy with anti-PD-L1 mAb therapy, whereas anti-PD-L2 mAb alone hardly induced any anti-tumor effects, suggesting that PD-L2’s function becomes evident when the effects of PD-L1 are abrogated by anti-PD-L1 mAb. Consistent with this notion, PD-L2 expression was upregulated on tumor-associated macrophages (TAM) when mice were treated with anti-PD-L1 mAb. Taken together, our study elucidated the importance of PD-L1 associated with tumor cells and non-tumor host cells, particularly BM-derived hematopoietic cells, as well as PD-L2 inducibly expressed on TAM in the suppression of anti-tumor immunity in the tumor microenvironment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immune checkpoint blockade therapies targeting programmed cell death-1 (PD-1) and programmed cell death-ligand 1 (PD-L1) have demonstrated significant clinical benefits superior to standard therapies in various types of advanced cancers, thus causing paradigm shift in the treatment of cancer [1,2,3]. In spite of the broad clinical applications, molecular and cellular mechanisms of PD-1/PD-L1 blockade are yet to be fully explored. In particular, it remains unknown which cells are targeted by PD-1/PD-L1 blockade therapy and how it modifies immune responses in the tumor microenvironment. Exploring these issues is highly important, as it could reveal novel biomarkers associated with positive responses to PD-1/PD-L1 blockade therapy and it may contribute to the identification of novel targets of cancer immunotherapy.
One crucial and unsolved issue is whether PD-L1 on tumor cells or non-tumor host cells is responsible for the suppressive functions in anti-tumor T-cell responses, and which of these cells are the primary targets of anti-PD-1/PD-L1 monoclonal antibodies (mAbs). In this regard, data from various clinical studies have demonstrated that PD-L1 expression level on tumor cells can be used as a predictive biomarker of patients who will display clinical benefits by these therapies [4, 5]. On the other hand, it has also been reported that PD-L1 positivity on tumor cells shows no correlation with therapeutic benefits of anti-PD-1 mAb [6]. Other studies have further indicated that PD-L1 expression on immune cells infiltrating in tumor tissues is associated with a higher response rate in patients treated with PD-1/PD-L1 blockade [7]. To experimentally investigate this issue, preclinical studies using PD-L1-deficient tumor cells and PD-L1-knockout (KO) mice have been conducted by several groups [8,9,10,11,12,13]. Inconsistent results have been reported in these studies, as crucial roles of PD-L1 on both tumor and non-tumor host cells [8,9,10], predominantly on tumor cells [11] or non-tumor host cells [12, 13] have been suggested. Currently, the relative importance of PD-L1 on tumor cells vs. non-tumor host cells in the suppression of anti-tumor immunity and in PD-1/PD-L1 blockade therapy remains controversial and thus warrants further investigation.
PD-1 delivers an inhibitory signal by interacting with programmed cell death-ligand 2 (PD-L2) as well as PD-L1 [14]. Therefore, another important question is whether PD-L2 expression in tumor tissues plays a role in the suppression of anti-tumor immunity and if so, how and what types of cells express PD-L2 in the tumor microenvironment. Although PD-L2 expression was initially thought to be restricted to macrophages and dendritic cells upon stimulation with IFN-γ, GM-CSF, or IL-4 [15], recent studies have indicated that various types of tumor cells and non-tumor host cells, including immune cells, endothelial cells, and fibroblasts, express PD-L2 [16,17,18]. Regarding the role of PD-L2 in the regulation of anti-tumor immunity, it remains controversial as one study demonstrated a significant correlation between PD-L2 expression and clinical benefits of anti-PD-1 mAb therapy [17], while another did not observe such correlation [19]. Thus, it is of importance to explore the potential roles of PD-L2 in suppressing of anti-tumor immunity and resistance to PD-1/PD-L1 blockade therapies.
In this study, we explored the relative importance of PD-L1 expressions on tumor cells and non-tumor host cells including bone marrow (BM)-derived hematopoietic cells in the regulation of anti-tumor immunity. In addition, the importance of PD-L2 expression on tumor-associated macrophages (TAM) as a biomarker and a therapeutic target for immune checkpoint blockade was investigated.
Materials and methods
Mice
PD-L1-KO mice, with a C57BL/6 background, were originally generated by Lieping Chen [20] (Yale University, CT). All mice were maintained under specific pathogen-free conditions in the animal facility at Yamaguchi University (Ube, Japan).
Tumor cell lines
3LL is a mouse lung carcinoma, B16F10 is a mouse melanoma, and MC38 is a mouse colon carcinoma cell line [21]. These cell lines were maintained in RPMI supplemented with 10% FBS, 1% penicillin–streptomycin, 25 mM HEPES, and 50 mM 2-mercaptoethanol.
Generation of a PD-L1-deficient MC38 cell line
To generate an MC38 cell line lacking PD-L1 expression, we designed a single-guide RNA targeting mouse PD-L1 (5′-TCCAAAGGACTTGTACGTGG-3′). Lentivirus particles (U6-gRNA/Puro-Cas9-GFP) were purchased from Sigma-Aldrich and transfected into MC38 cells in the presence of polybrene (hexadimethrine bromide, Sigma-Aldrich). After puromycin selection, a single cell of GFP-positive MC38 was isolated by FACS (SH800, Sony). A control MC38 cell line was generated in the same manner using CRISPR-Lenti Non-Targeting Control Transduction Particles (Sigma-Aldrich). Expression levels of PD-L1/L2 and MHC class I/II on these cell lines were examined by flow cytometry (BD LSRFortessa X-20, BD Biosciences) after staining with the following reagents; PE-conjugated anti-PD-L1 mAb (clone M1H5, eBioscience), PE-conjugated anti-H-2Kb mAb (clone AF6-88.5, BD Biosciences), PE-conjugated anti-H-2Db mAb (clone KH95, Biolegend), PE-conjugated anti-I-Ab mAb (clone AF6-120.1, BD Biosciences), and APC-conjugated anti-PD-L2 mAb (clone TY25, BD Biosciences). In some experiments, MC38 cell lines were incubated in the presence of mouse IFN-γ (Biolegend) prior to the analyses.
In vivo tumor models
Wild-type C57BL/6 mice or PD-L1-KO mice were inoculated subcutaneously (s.c.) with 1 × 106 PD-L1-deficient or control MC38 in the right lateral flank. In some experiments, wild-type C57BL/6 mice were inoculated s.c. with 1 × 106 3LL tumor cells. These tumor cells were suspended in HBSS prior to injection. For Ab treatment, 200 µg of hamster IgG (Innovative Research), rat IgG (Sigma-Aldrich), anti-mouse PD-L1 mAb (clone 10B5) [22], or anti-mouse PD-L2 mAb (clone TY25, purchased from BioXcell) were injected intraperitoneally (i.p.) on days 4, 9, 14, 19, and 24 after tumor inoculation. Tumor growth was measured at least twice a week with a digital caliper and tumor volume was calculated using the following formula; x × y2/2, where x is the long diameter and y is the short diameter of the tumor. Mice were euthanized when tumor volume reached 4000 mm3 or severe ulceration with bleeding in the tumor was observed.
Bone marrow chimeric mice
BM cells were harvested from wild-type or PD-L1-KO mice by flushing the marrow cavity of the femur with RPMI medium. After lysis of red blood cells with Ammoniumm–Chloride–Potassium Lysing Buffer (Lonza), BM cells were suspended with HBSS, passed through a cell strainer, and counted. Recipient wild-type or PD-L1-KO mice were given a lethal dose of irradiation, consisting of two split 6 Gy doses, 6–8 h apart (total of 12 Gy), followed by intravenous injection of 7.5 × 106 BM cells per mouse. At least 6 weeks later, BM chimeric mice were inoculated s.c. with 1 × 106 PD-L1-deficient MC38 cells.
Analysis of tumor tissue by flow cytometry
Wild-type C57BL/6 mice were inoculated s.c. with MC38 and treated with hamster IgG or anti-PD-L1 mAb on day 4. On day 9, tumor tissue was resected and minced with scissors, followed by digestion with medium containing liberase TL (Roche) and DNaseI (Roche) for 2 h at room temperature. Digested tumor samples were homogenized by repetitive pipetting and passed through cell strainers to generate single-cell suspensions. Tumor-infiltrating immune cells were separated from tumor and stromal cells by magnetic cell sorting using anti-mouse CD45 mAb (Miltenyi Biotec). CD45-negative populations were stained with APC-conjugated anti-PD-L2 mAb (clone TY25, BD Biosciences). CD45-positive populations were stained with the following mAbs; BV421-conjugated anti-CD11b mAb (clone M1/70, BD Biosciences), PE-Cy7-conjugated anti-F4/80 mAb (clone BM8, Biolegend), and APC-conjugated anti-PD-L2 mAb (clone TY25, BD Biosciences). TAM were identified as a population double positive for CD11b and F4/80 within CD45-positive cells, while the remaining populations (i.e., CD11b and F4/80 single positive or double negative) within CD45-positive cells were identified as non-TAM immune cells, including T cells. Expression levels of PD-L2 on cells of tumor tissues were examined by flow cytometer (BD LSRFortessa X-20), and the data were analyzed using FlowJo software (FlowJo, LLC). All cells were pre-incubated with anti-CD16/32 mAb (Fc block, clone 2.4G2, BD Biosciences) prior to staining to block non-specific binding of mAbs.
Rechallenge of tumor
Mice which had rejected the MC38 tumor after combined treatment with anti-PD-L1 and anti-PD-L2 mAbs were rechallenged s.c. with MC38 and B16F10 in the right and left lateral flank, respectively, 3 months after the original tumor inoculation. As a control, naïve C57BL/6 mice were also inoculated s.c. with MC38 and B16F10 in the same manner. Tumor growth was measured at least twice a week with a digital caliper.
Statistical analysis
JMP 13 (SAS Institute Inc., Cary, NC) was used for statistical analysis. The two-tailed Student’s t test was applied to compare two groups. For survival data, Kaplan–Meier survival curves were prepared and statistical differences were analyzed using the log-rank test. p < 0.05 was considered statistically significant.
Results
PD-L1 on tumor cells and non-tumor cells mediates suppression of anti-tumor immune responses
Expressions of PD-L1 in the tumor microenvironment are detectable on both tumor cells and non-tumor cells including stromal and infiltrating immune cells. First, to explore the relative importance of PD-L1 expressed on tumor vs. non-tumor cells, we developed a model in which PD-L1-deficient or competent tumor cells were inoculated into PD-L1-KO or wild-type mice. PD-L1-deficient tumor cells were generated from MC38 mouse colon carcinoma by a CRISPR/Cas9 gene-editing method. PD-L1-deficient MC38 did not express PD-L1, even in the presence of IFN-γ, while control MC38 treated with a scrambled CRISPR/Cas9 gRNA showed a significant upregulation of PD-L1 in response to IFN-γ (Supplementary Fig. 1a). We also confirmed that expression levels of MHC class I were comparable between PD-L1-deficient and control MC38 cell lines (Supplementary Fig. 1b). The extent of MHC class I upregulation by stimulation with IFN-γ was also equivalent in these cell lines. Neither PD-L2 nor MHC class II were expressed on these cell lines in the presence or absence of IFN-γ (Supplementary Fig. 1a, b).
To explore importance of PD-L1 expressed on tumor cells or non-tumor cells in the suppression of anti-tumor immunity, PD-L1-deficient or control MC38 cells were s.c. injected into PD-L1-KO or wild-type mice. While control MC38 grew in all cases when inoculated into wild-type mice, PD-L1-deficient MC38 showed a significant delay in tumor growth and resulted in tumor rejection in 6 out of 15 mice (Fig. 1a). Survival was also significantly prolonged in wild-type mice inoculated with PD-L1-deficient MC38 (Fig. 1b). When control MC38 cells were inoculated into PD-L1-KO mice, tumor rejection was observed in 1 out of 17 mice along with a delay in tumor growth and prolonged mouse survival. Importantly, when PD-L1-deficient MC38 cells were inoculated into PD-L1-KO mice, complete tumor rejection and long-term survival were observed in 16 out of 17 mice. Taken together, these results suggest that PD-L1 on tumor cells and non-tumor host cells are both important in the suppression of anti-tumor immunity, while PD-L1 on tumor cells makes a primary contribution.

Important role of PD-L1 on tumor and host cells in anti-tumor immune responses. Control or PD-L1-deficient MC38 tumor cells were inoculated s.c. into wild-type or PD-L1-KO mice. a Tumor growth in each group is shown. Each line indicates the tumor size in individual mice. Data are shown from two independent experiments. The number of tumor-rejected mice out of the total number of mice is indicated. b Mouse survival rates are shown. Open circle: wild-type mice inoculated with control MC38, open triangle: wild-type mice inoculated with PD-L1-deficient MC38, filled circle: PD-L1-KO mice inoculated with control MC38, filled triangle: PD-L1-KO mice inoculated with PD-L1-deficient MC38. Open circle vs. filled circle; p = 0.0005, open circle vs. open triangle; p < 0.0001, filled circle vs. open triangle; p = 0.025, open triangle vs. filled triangle; p = 0.0006
Important role of PD-L1 on bone marrow-derived hematopoietic cells in the suppression of anti-tumor immunity
Our data revealed a potential role of PD-L1 expressed on non-tumor host cells in the suppression of anti-tumor immunity. However, it remains unclear whether stromal non-immune cells or infiltrating immune cells are responsible for this effect, since PD-L1 can be detected on various host cells, including endothelial cells and cancer-associated fibroblasts [18, 23]. To address this question, BM chimeric mice, in which wild-type mice were treated with systemic myeloablative irradiation followed by transferring BM cells from PD-L1-KO mice (PD-L1-KO BM into wild-type mice), were generated. In addition, mice with BM transfer vice versa (wild-type BM into PD-L1-KO mice) were also generated. These BM chimeric mice were inoculated s.c. with PD-L1-deficient MC38. In PD-L1-KO BM into wild-type mice, tumors were rejected in all cases (Fig. 2). On the other hand, eventual tumor growth was observed in four out of ten cases in wild-type BM into PD-L1-KO mice. These results suggest that, among non-tumor host cells, PD-L1 expressed on BM-derived hematopoietic cells, including immune cells, plays a major role in the suppression of anti-tumor immunity.

Suppressive role of PD-L1 on BM-derived hematopoietic cells in anti-tumor immunity. BM chimeric mice, generated by transferring PD-L1-KO BM cells into wild-type mice or vice versa, were inoculated s.c. with PD-L1-deficient MC38. a Tumor growth in each group is shown. Each line indicates the tumor size in individual mice. The number of tumor-rejected mice out of the total number of mice is indicated. b Mouse survival rates are shown. Open circle: PD-L1-KO BM transferred into wild-type mice, filled triangle: wild-type BM transferred into PD-L1-KO mice. Open circle vs. filled triangle; p = 0.0383
Anti-tumor effects of PD-L1 and PD-L2 blockade in MC38 tumor model
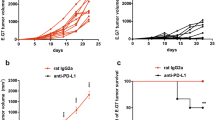
The inhibitory effects of PD-1 can be mediated by its interaction with PD-L2 as well as PD-L1 [14]. To explore the potential role of PD-L2 in the suppression of anti-tumor immunity and its relevance to PD-L1 functions, mice inoculated with MC38 were treated with anti-PD-L2 mAb with or without anti-PD-L1 mAb. As control, rat IgG and hamster IgG were injected, respectively. Treatment with anti-PD-L2 mAb alone induced hardly any anti-tumor effects, as shown that tumor growth and mouse survival were equivalent to those treated with control Abs (Fig. 3). On the other hand, treatment with anti-PD-L1 mAb alone significantly inhibited tumor growth and induced tumor rejection in 5 out of 12 mice, resulting in prolonged mouse survival. When anti-PD-L2 mAb was injected in combination with anti-PD-L1 mAb, tumor growth was further inhibited and resulted in tumor rejection in 11 out of 12 mice. Survival of mice treated with both anti-PD-L1 and anti-PD-L2 mAbs was significantly prolonged compared to those treated with anti-PD-L1 mAb alone. These results suggest that the suppressive effects of PD-L2 are undetectable by itself, but become evident under conditions that PD-L1/PD-1 interaction is ablated.

Therapeutic effects of anti-PD-L1 and anti-PD-L2 mAbs in MC38. Wild-type mice were inoculated s.c. with MC38 and treated with anti-PD-L1 mAb alone, anti-PD-L2 mAb alone, or a combination of these mAbs. Hamster IgG and rat IgG were used as control Abs. a Tumor growth in each group is shown. Each line indicates the tumor size in individual mice. Data are shown from two independent experiments. The number of tumor-rejected mice out of the total number of mice is indicated. b Mouse survival rates are shown. Open circle: control Abs, open triangle: anti-PD-L1 mAb + control Ab, filled circle: anti-PD-L2 mAb + control Ab, filled triangle: anti-PD-L1 mAb + anti-PD-L2 mAb. Open circle vs. filled circle; p = 0.121, open circle vs. open triangle; p < 0.0001, open triangle vs. filled triangle; p = 0.011
Enhanced expression of PD-L2 on tumor-associated macrophages by PD-L1 blockade
To investigate the mechanism by which the effects of PD-L2 become evident along with PD-L1 blockade, expression levels of PD-L2 on cells in the tumor microenvironment, including tumor cells and infiltrating immune cells, were analyzed in the presence or absence of anti-PD-L1 mAb treatment. Mice were inoculated with MC38 on day 0 and then treated with anti-PD-L1 mAb or control Ab on day 4. On day 9, tumor tissue was harvested and digested to a single-cell suspension, followed by separation into CD45-positive immune cells and CD45-negative non-immune cells by magnetic sorting. Expression levels of PD-L2 were assessed by flow cytometry, in which TAM were identified as CD45+CD11b+F4/80+ cells, while the remaining CD45+ subsets, i.e., CD11b, F4/80 single positive or double negative, were considered to be non-TAM immune cells, including T cells, B cells, NK cells, and dendritic cells. In the absence of anti-PD-L1 mAb treatment, slight expressions of PD-L2 were detected on TAM, but not other CD45+ subsets (Fig. 4). When mice were treated with anti-PD-L1 mAb, PD-L2 expression on TAM, but not other CD45+ subsets, significantly increased. There were no significant differences in the number of CD45-positive immune cells in the tumor tissue or the percentage of TAM between control Ab- and anti-PD-L1 mAb-treated groups (data not shown). No expression of PD-L2 was detected on CD45-negative non-immune cells, which included tumor cells, irrespective of treatment with anti-PD-L1 mAb. These results reveal that PD-L2 expression is inducibly upregulated on TAM in the presence of PD-L1 blockade.

Inducible expression of PD-L2 on TAM by anti-PD-L1 mAb treatment. Wild-type mice were inoculated s.c. with MC38 and treated with anti-PD-L1 mAb or control Ab. Tumor tissue was harvested and analyzed for the expression of PD-L2 on TAM, non-TAM immune cells, and CD45-negative non-immune cells by flow cytometry. a Representative histograms are shown. The filled and solid lines indicate unstained controls and stained samples, respectively. b Percentages of PD-L2 positive cells in TAM, non-TAM immune cells, and CD45-negative non-immune cells were analyzed. Data are shown as mean ± SEM of ten or eight mice per group. Data are shown from two independent experiments. **p = 0.0054, NS not significant
Long-term anti-tumor memory responses induced by treatment with anti-PD-L1 and anti-PD-L2 mAbs
Combined treatment with anti-PD-L1 and anti-PD-L2 mAbs achieved MC38 tumor rejection in almost all mice, resulted in prolonged survival over 100 days. To confirm the generation of tumor-specific memory responses with this treatment, the survived mice were rechallenged with MC38 and B16F10, melanoma cells syngeneic to C57BL/6 mice but unrelated to MC38 in terms of antigenicity. It was found that all the tumor-survived mice were resistant to rechallenge with MC38 but not B16F10 (Fig. 5). As a control, MC38 and B16F10 inoculated into naïve C57BL/6 mice in the same manner led to apparent tumor growth. This result indicates that combined blockade of PD-L1 and PD-L2 can induce tumor-specific long-term memory responses.

Induction of tumor-specific memory response by treatment with anti-PD-L1 and anti-PD-L2 mAbs. Mice inoculated with MC38 were treated with both anti-PD-L1 and anti-PD-L2 mAbs to induce tumor regression. After 3 months, the tumor-rejected mice (open circle) were rechallenged s.c. with MC38 and B16F10 on the right and left lateral flank, respectively. As a control, naïve C57BL/6 mice (filled circle) were also inoculated s.c. with MC38 and B16F10 in the same manner. Tumor sizes were measured and are shown as the mean ± SD of five or six mice per group
Therapeutic effects of anti-PD-L1 and anti-PD-L2 mAbs in 3LL lung tumors
The therapeutic effects of combined treatment with anti-PD-L1 and anti-PD-L2 mAbs were further examined in another tumor model using 3LL, Lewis lung carcinoma. Mice inoculated s.c. with 3LL tumor were treated with anti-PD-L1 mAb, anti-PD-L2 mAb, or both of these mAbs. As shown in Fig. 6, combined therapy of anti-PD-L1 and anti-PD-L2 mAbs significantly inhibited 3LL tumor growth, resulting in prolonged mouse survival compared with the other groups (p = 0.013 vs. control Abs, p = 0.021 vs. anti-PD-L1 mAb, p = 0.021 vs. anti-PD-L2 mAb). No significant differences in survival were observed among other groups. This result indicates that the synergistic anti-tumor effects of simultaneous blockade of PD-L1 and PD-L2 are also detectable in a 3LL lung tumor model.

Combined treatment with anti-PD-L1 and anti-PD-L2 mAbs in a 3LL lung tumor model. Mice were inoculated s.c. with 3LL and treated with anti-PD-L1 mAb alone, anti-PD-L2 mAb alone, or a combination of these mAbs. Hamster IgG and rat IgG were used as control Abs. a Tumor growth in each group is shown as the mean ± SD of five mice per group. *: p < 0.05. b Mouse survival rates are shown. Open circle: control Abs, open triangle: anti-PD-L1 mAb + control Ab, filled circle: anti-PD-L2 mAb + control Ab, filled triangle: anti-PD-L1 mAb + anti-PD-L2 mAb. open circle vs. filled triangle; p = 0.013, open triangle vs. filled triangle; p = 0.021, filled circle vs. filled triangle; p = 0.021
Discussion
In this study, we attempted to elucidate the molecular and cellular mechanisms by which PD-L1 and PD-L2 inhibit anti-tumor T-cell responses in the tumor microenvironment. Our findings indicate that PD-L1 on both tumor cells and non-tumor host cells mediates inhibitory effects, while tumor-associated PD-L1 plays a predominant role. Among non-tumor host cells, PD-L1 on BM-derived hematopoietic cells was found to be essential. Although PD-L2 mediated almost no effects in the presence of the PD-L1/PD-1 interaction, its immune-inhibitory effects became evident, through inducible expression on TAM, when the PD-L1/PD-1 interaction was attenuated. These findings provide useful insights into the clinical applications of PD-1/PD-L1 blockade therapies regarding the identification of accurate biomarkers and the development of efficient immunotherapies.
Several previous studies have explored the importance of PD-L1 expressed on tumor cells and non-tumor host cells utilizing PD-L1-deficient tumor lines and/or PD-L1-KO mice [8,9,10,11,12,13]. While these studies have reached inconsistent observations, i.e., crucial roles of PD-L1 on both tumor and non-tumor cells, predominantly on tumor cells, or host cells, these results are probably due to differences in experimental models, including the immunogenicity of tumors and injection doses of cells and reagents. In this regard, our study indicated that both tumor- and host-derived PD-L1 can inhibit anti-tumor immune responses. It should be noted that our findings further indicated more primary role of PD-L1 on tumor cells than that on host cells, based on direct comparison between wild-type mice inoculated with PD-L1-deficient tumors and PD-L1-KO mice inoculated with control tumors. Among host cells, PD-L1 associated with BM-derived hematopoietic cells, but not non-hematopoietic cells, played an essential role in the suppression of anti-tumor immune responses. This is consistent with previous studies indicating the importance of PD-L1 on macrophages and dendritic cells [12, 13]. These preclinical studies collectively suggest that PD-L1 on tumor cells and host hematopoietic cells are both involved in suppressing anti-tumor T-cell immunity, while their relative importance changes depending on various factors, including tumor immunogenicity and endogenous expression of PD-L1 by genetic and/or epigenetic control. Further studies utilizing clinical samples from various cancers are required to fully explore the role of PD-L1 in the tumor microenvironment.
While the importance of PD-L2 as a target and potential biomarker of anti-PD-1 mAb therapy has been suggested [17], the precise mechanisms how PD-L2 inhibits T-cell immunity in the tumor microenvironment have remained unexplored. Our findings in this study revealed that PD-L2 expression is upregulated on TAM and its inhibitory effects become evident when PD-L1 function is abrogated by anti-PD-L1 mAb. This result implies that, although PD-L1/PD-1-dependent suppression is the primary mechanism of immune evasion in cancer, alternative mechanisms that include PD-L2 upregulation, may compensate once PD-L1 function is dampened. These findings are consistent with previous reports suggesting that the presence of TAM correlates with poor prognosis in human cancers [24] and that PD-L2 is expressed on non-tumor cells according to tumor cell types and the conditions of the tumor microenvironment [17]. Regarding the molecular mechanisms how PD-L2 expression is induced by PD-L1 blockade, it has been reported that PD-L2 on TAM is upregulated by IL-27 via Stat3 activation [25]. While the detailed mechanism of the PD-L2 upregulation in our study remains unclear, we infer that changes in the cytokine milieu in the tumor microenvironment by anti-PD-L1 mAb treatment may trigger the expression of PD-L2. IFN-γ may play a certain role, since IL-27 production by macrophages can be induced by IFN-γ-mediated pathways [26].
In addition to PD-L2, various inhibitory mechanisms, including PD-1-independent immune checkpoint molecules, regulatory T cells, and suppressive cytokines/enzymes, may also mediate the compensatory effects when the PD-L1/PD-1 system is abrogated. Consistent with this notion, upregulation of TIM-3 in response to anti-PD-1 mAb treatment has been reported [27]. Furthermore, combined therapy of anti-PD-1 mAb with anti-TIM-3, LAG-3, or TIGIT mAb induces remarkable synergy to enhance the anti-tumor effects of anti-PD-1 mAb, whereas monotherapy of anti-TIM-3, LAG-3, or TIGIT mAb hardly displays any therapeutic potential [28,29,30], suggesting that these checkpoint molecules become adaptively functional following PD-1 blockade. Taken together, adaptive resistance of a tumor is a highly dynamic process which can be affected by endogenous T-cell responses, as well as exogenous medical intervention, including immunotherapies. Serial evaluation of immune-regulatory molecules before and after immunotherapies is necessary for the development of effective combination immunotherapies and the identification of highly predictive biomarkers.
Abbreviations
- ATCC:
-
American Type Culture Collection
- BM:
-
Bone marrow
- i.p.:
-
Intraperitoneally
- KO:
-
Knockout
- mAb:
-
Monoclonal antibody
- mAbs:
-
Monoclonal antibodies
- PD-1:
-
Programmed cell death-1
- PD-L1:
-
Programmed cell death-ligand 1
- PD-L2:
-
Programmed cell death-ligand 2
- s.c.:
-
Subcutaneously
- TAM:
-
Tumor-associated macrophages
References
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366(26):2443–2454. https://doi.org/10.1056/NEJMoa1200690
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366(26):2455–2465. https://doi.org/10.1056/NEJMoa1200694
Ribas A, Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359(6382):1350–1355. https://doi.org/10.1126/science.aar4060
Garber K (2015) Predictive biomarkers for checkpoints, first tests approved. Nat Biotechnol 33(12):1217–1218. https://doi.org/10.1038/nbt1215-1217
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, O’Brien M, Rao S, Hotta K, Leiby MA, Lubiniecki GM, Shentu Y, Rangwala R, Brahmer JR (2016) Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 375(19):1823–1833. https://doi.org/10.1056/NEJMoa1606774
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P, CheckMate I (2015) Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 373(19):1803–1813. https://doi.org/10.1056/NEJMoa1510665
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, Joseph RW, Galsky MD, Fleming MT, Petrylak DP, Perez-Gracia JL, Burris HA, Castellano D, Canil C, Bellmunt J, Bajorin D, Nickles D, Bourgon R, Frampton GM, Cui N, Mariathasan S, Abidoye O, Fine GD, Dreicer R (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387(10031):1909–1920. https://doi.org/10.1016/S0140-6736(16)00561-4
Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, Selby MJ, Graziano RF, Mardis ER, Korman AJ, Schreiber RD (2017) Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res 5(2):106–117. https://doi.org/10.1158/2326-6066.CIR-16-0391
Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, Sanders L, Koeppen H, Caplazi P, McBride J, Chiu H, Hong R, Grogan J, Javinal V, Yauch R, Irving B, Belvin M, Mellman I, Kim JM, Schmidt M (2017) Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun 8:14572. https://doi.org/10.1038/ncomms14572
Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF (2017) PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology 6(4):e1294299. https://doi.org/10.1080/2162402X.2017.1294299
Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH (2017) PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 214(4):895–904. https://doi.org/10.1084/jem.20160801
Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, Liu X, Harrington SM, Guo J, Xin Y, Xiong Y, Nham K, Silvers W, Hao G, Sun X, Chen M, Hannan R, Qiao J, Dong H, Peng H, Fu YX (2018) PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Investig 128(2):580–588. https://doi.org/10.1172/JCI96061
Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, Vats P, Chinnaiyan AM, Lao CD, Lawrence TS, Wicha M, Hamanishi J, Mandai M, Kryczek I, Zou W (2018) Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Investig 128(2):805–815. https://doi.org/10.1172/JCI96113
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2(3):261–268. https://doi.org/10.1038/85330
Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H (2002) Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169(10):5538–5545. https://doi.org/10.4049/jimmunol.169.10.5538
Messal N, Serriari NE, Pastor S, Nunes JA, Olive D (2011) PD-L2 is expressed on activated human T cells and regulates their function. Mol Immunol 48(15–16):2214–2219. https://doi.org/10.1016/j.molimm.2011.06.436
Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J, Lunceford J, Cheng J, Chow LQM, Seiwert TY, Handa M, Tomassini JE, McClanahan T (2017) PD-L2 expression in human tumors: relevance to anti-PD-1 therapy in cancer. Clin Cancer Res 23(12):3158–3167. https://doi.org/10.1158/1078-0432.CCR-16-1761
Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ, Yokota SJ, Bankert RB (2007) Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol 178(9):5552–5562. https://doi.org/10.4049/jimmunol.178.9.5552
Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 20(19):5064–5074. https://doi.org/10.1158/1078-0432.ccr-13-3271
Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L (2004) B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity 20(3):327–336
Clarke P, Mann J, Simpson JF, Rickard-Dickson K, Primus FJ (1998) Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res 58(7):1469–1477
Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L (2005) Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 65(3):1089–1096
Mazanet MM, Hughes CCW (2002) B7-H1 Is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol 169(7):3581–3588. https://doi.org/10.4049/jimmunol.169.7.3581
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4(1):71–78. https://doi.org/10.1038/nrc1256
Horlad H, Ma C, Yano H, Pan C, Ohnishi K, Fujiwara Y, Endo S, Kikukawa Y, Okuno Y, Matsuoka M, Takeya M, Komohara Y (2016) An IL-27/Stat3 axis induces expression of programmed cell death 1 ligands (PD-L1/2) on infiltrating macrophages in lymphoma. Cancer Sci 107(11):1696–1704. https://doi.org/10.1111/cas.13065
Liu J, Guan X, Ma X (2007) Regulation of IL-27 p28 gene expression in macrophages through MyD88- and interferon-gamma-mediated pathways. J Exp Med 204(1):141–152. https://doi.org/10.1084/jem.20061440
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong KK, Hammerman PS (2016) Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7:10501. https://doi.org/10.1038/ncomms10501
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC (2010) Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 207(10):2187–2194. https://doi.org/10.1084/jem.20100643
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72(4):917–927. https://doi.org/10.1158/0008-5472.CAN-11-1620
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, Eaton DL, Grogan JL (2014) The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26(6):923–937. https://doi.org/10.1016/j.ccell.2014.10.018
Acknowledgements
The authors thank Shunsuke Goto, Hiromi Kurosawa and Makiko Miyamoto for excellent technical assistance.
Funding
This study was supported by research funds from Grant-in-Aid for Scientific Research 16H02474 and Ono Pharmaceutical Inc.
Author information
Authors and Affiliations
Contributions
DU, NO, YS, and KA conducted experiments. TO, HY, ME, and KT guided the conduct of experiments. DU and KT wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Koji Tamada received research funds from Ono Pharmaceutical Inc. Other authors declare no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted (Yamaguchi University, Ube, Japan). Animal research was approved by the Institutional Animal Care and Use Committee of Yamaguchi University (animal research approval number: 14-001).
Animal source
Male or female 6 to 12-week-old wild-type C57BL/6 mice were purchased from Japan SLC (Shizuoka, Japan). PD-L1-KO mice with a C57BL/6 background were kindly provided by Lieping Chen.
Cell line authentication
The MC38 mouse colon carcinoma cell line was kindly provided by F. James Primus. The 3LL mouse lung carcinoma cell line and the B16F10 mouse melanoma cell line were purchased from Japanese Collection of Research Bioresources Cell Bank and American Type Culture Collection (ATCC), respectively, who had authenticated them.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Umezu, D., Okada, N., Sakoda, Y. et al. Inhibitory functions of PD-L1 and PD-L2 in the regulation of anti-tumor immunity in murine tumor microenvironment. Cancer Immunol Immunother 68, 201–211 (2019). https://doi.org/10.1007/s00262-018-2263-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-018-2263-4