
Abstract
The construction of a tumor-associated carbohydrate antigen-zwitterionic polysaccharide conjugate, Thomsen-nouveau-polysaccharide A1 (Tn-PS A1, where Tn = d-GalpNAc), has led to the development of a carbohydrate binding monoclonal antibody named Kt-IgM-8. Kt-IgM-8 was produced via hybridoma from Tn-PS A1 hyperimmunized Jackson Laboratory C57BL/6 mice, splenocytes and the murine myeloma cell line Sp2/0Ag14 with subsequent cloning on methyl cellulose semi-solid media. This in-house generated monoclonal antibody negates binding influenced from peptides, proteins, and lipids and preferentially binds monovalent Tn antigen as noted by ELISA, FACS, and glycan array technologies. Kt-IgM-8 demonstrated in vitro and in vivo tumor killing against the Michigan Cancer Foundation breast cell line 7 (MCF-7). In vitro tumor killing was observed using an LDH assay that measured antibody-induced complement-dependent cytotoxicity and these results were validated in an in vivo passive immunotherapy approach using an MCF-7 cell line-derived xenograft model. Kt-IgM-8 is effective in killing tumor cells at 30% cytotoxicity, and furthermore, it demonstrated approximately 40% reduction in tumor growth in the MCF-7 model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A cancer antigen prioritization study revealed 9 out of the top 75 cancer antigens as tumor-associated carbohydrate antigens (TACAs) [1]. Carbohydrate tumor antigens are viable targets for the development of immunotherapies, in which, there are many TACA-based vaccines currently being evaluated [2,3,4,5,6]. Recently, the Food and Drug Administration (FDA) approved Unituxin® (dinutuximab) from a National Cancer Institute (NCI)/United Therapeutics joint venture as the first mAb to target the TACA ganglioside disialic acid 2 (GD2 = d-GalpNAcβ1–4(Neu5Acα2–8Neu5Acα2–3)-d-Galpβ1–4Glc) for the treatment of high-risk neuroblastomas in pediatric patients [7]. Prior to Unituxin®, the FDA approved therapeutic mAbs which targeted protein-based tumor antigens such as Avastin® (bevacizumab—Genentech, Inc.), Herceptin® (trastuzumab—Genentech, Inc.), and Rituxan® (rituximab—Biogen Idec Inc.) [8, 9].
Unlike proteins, TACAs elicit a T-cell independent immune response resulting in weak immunogenicity. This limitation is alleviated when TACAs are conjugated to immunogenic carrier proteins. This approach has some disadvantages due to protein-epitope suppression and non-specific antibody binding caused by immunogenic hydrocarbon linkers [10,11,12]. There is often ambiguity in the effectiveness of TACA conjugates as vaccines and there is a poor record of Phase III clinical trials including THERATOPE® (sialyl Thomsen-nouveau-keyhole limpet hemocyanin (STn-KLH conjugate)) [13, 14]. The current protein immunogen carrier strategy to target carbohydrate antigens for eradicating cancer could be improved by investigating alternative immunogenic carriers.
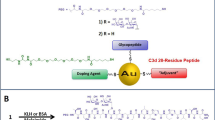
One approach for overcoming issues of TACA-protein conjugates and increasing carbohydrate immunogenicity is the use of an entirely carbohydrate-based immunogen such as Thomsen-nouveau-polysaccharide A1 (Tn-PS A1) (1) (Fig. 1). Tn-PS A1 (1) features the Tn (d-GalpNAc) antigen and an MHCII-binding zwitterionic polysaccharide (ZPS), PS A1, isolated from the capsule of Bacteroides fragilis [American-Type Culture Collection (ATCC) 25285/National Collection of Type Cultures (NCTC) 9343] [15]. Conjugate 1, derived from oxidized PS A1 and a synthetic aminooxy Tn derivative capitalizing on oxime formation, is stable under physiological conditions [15, 16]. This unique immunogen stimulates anti-tumor responses through the induction of CD4+ T cells polarized by cytokines IL-2, IL-4, IL-10, and IL-17A encoding for Th1/Th17 immunity [17, 18]. Furthermore, carbohydrate-selective polyclonal IgG (pIgG) and polyclonal IgM (pIgM) antibodies have been observed [15, 17]. This innovative design for an entirely carbohydrate immunogen, capable of augmenting the immune response towards TACAs, may become a valuable platform for treating/preventing cancers when immunotherapeutic approaches are applied.

Structure of Tn-PS A1 (1)
Although many FDA approved antibodies are monoclonal IgGs (mIgGs), there have been ongoing clinical investigations using monoclonal IgMs (mIgMs) such as mAb216 and L612 HuMAb, which have demonstrated promising therapeutic results in melanoma and leukemia patients respectively [19, 20]. IgM antibodies are gaining clinical relevance due in part to their pentavalent nature and increased ability to initiate complement-dependent cytotoxicity (CDC) [21,22,23]. The pentavalent structure allows for high avidity due to increased antigen-binding events. In addition, IgM antibodies allow for enhanced CDC activity when compared to their IgG counterparts due to the initial complement protein, C1q, which binds 103-fold greater to Fc-IgM. C1q is involved in the initial C1 complex of the classical complement cascade [24, 25]. Thus, by targeting TACAs using mIgM antibodies, there is an increased therapeutic potential through multivalent target avidity and complement activation for immunotherapy.
Other mAbs (IgG and IgM) have been produced to recognize TACAs, but unfortunately, many lack the ability to bind monovalent glycans and are limited to binding clustered or multivalent presentations of glycans [26]. In 2007, Gildersleeve and coworkers examined the binding events of 27 carbohydrate specific mAbs to various TACAs employing glycan array technology [26]. The results were particularly concerning due to the lack of binding to the cognate monovalent antigens, but rather, binding was observed with antigen clusters. Specifically, B1.1 (IgM), a commercial mAb against Tn failed to bind Tn alone, but rather was found to bind Tn antigens in clusters containing two or more GalNAcα1-Ser/Thr (Tn) [26]. Therefore, devising alternative strategies to address mAb binding to monovalent TACAs is a challenging, but critical endeavor for expanding the currently available glycan tool box.
An important criterion for producing anti-carbohydrate mAbs is that immunogens generate selective antibodies against sugar antigens without binding preference from immunogen peptide/hydrocarbon linkers. To limit mAb cross reactivity, we sought to utilize Tn-PS A1 (1) with a focus of immunity against the Tn antigen. To accomplish our goal, we have capitalized on knowledge previously gained by examining PS A1-based conjugates using an oxime bond providing a unique, entirely carbohydrate immunogen in the absence of artificial linkers. We utilized a synthetically prepared anomeric aminooxy Tn moiety to conjugate to oxidized PS A1 through an embedded aldehyde on the d-furanose moiety (Fig. 1) [15, 17]. This strategy places an emphasis on the immune response towards O-linked carbohydrates employing linker-free oxime ligation and not on O-linked glycopeptides. For example, mAbs that bind selectively towards glycopeptides tend to be influenced by the original peptide sequence and are thus not entirely glycan specific [27,28,29,30]. Traditional methods for mAb production have used naturally occurring entities that contain TACAs (i.e., cancer cells and glycosylated proteins) which have led to a plethora of non-selective commercially available mAbs such as B1.1 (mIgM) and Tn 218 (mIgM) which preferentially bind clustered Tn (epitopes with two or more Tn consecutive antigens) [31, 32]. B1.1 and Tn 218 were produced from ovine submaxillary mucin and screened for Tn binding. The complications associated with raised mAbs using glycoproteins include epitopic suppression due to increased immunogenicity towards the protein which minimizes the response against the target antigenic sugar [33, 34]. Most mAbs generated from glycopeptides/proteins/linkers have a varying degree of sensitivity towards the peptide/linker portion. However, our design concept uses an entirely carbohydrate immunogen providing a carbohydrate-selective mAb development process. Here, within, we highlight our Tn-PS A1 (1) construct that was used to produce mIgM antibodies where the donor/acceptor Fab/antigen-binding events are preferential for the Tn antigen. Furthermore, we demonstrate that the monoclonal antibody, termed Kt-IgM-8, possesses tumor killing activity against Michigan Cancer Foundation breast cell line 7 (MCF-7) in an LDH complement-dependent cytotoxicity assay and in an in vivo MCF-7 tumor xenograft model. We also demonstrate that the use of Tn-PS A1 generate mAbs capable of eliciting anti-tumor responses superior to some that are commercially available.
Materials and methods
Immunizations
Immunization of Tn-PS A1, PS A1, and PBS has been reported [15].
Hybridoma fusion protocol
Mouse spleens were obtained on day 60 and put in DMEM media. The splenocytes were obtained by homogenizing the spleens. Cells were washed with serum-free DMEM by centrifuging at 1000 rpm for 10 min and resuspending the final pellet in 30 mL of serum-free DMEM. Simultaneously, Sp2/0-Ag14 (ATCC CRL-1581) were cultured and washed with serum-free DMEM by centrifuging at 1000 rpm for 10 min followed by resuspension in 30 mL of serum-free DMEM. 2 × 107 myeloma cells and 1 × 108 viable splenocytes were added in a 50 mL centrifuge tube and were washed with serum-free DMEM three times. ClonaCell™-HY PEG (1 mL) was added without stirring. Cells were stirred for 1 min by gently agitating the tube. 4 mL of serum-free DMEM media was added to the fusion mixture and stirred for 4 min. 10 mL of serum-free DMEM was slowly added and the entire mix was incubated at 37 °C for 15 min. 30 mL of 10% FCS-DMEM (10-DMEM) was added and washed with 40 mL of DMEM and the supernatant was discarded. 10 mL of 20% FCS-DMEM (20-DMEM) was used to resuspend the pellet and was transferred to a T-175 flask containing 20 mL of 20-DMEM. The resuspended pellet was then incubated for 24 h in 5% CO2. Cells were centrifuged and resuspended with 10 mL of 20-DMEM and then added to 90 mL of semi-solid methyl cellulose media (ClonaCell™ FLEX). The bottle was mixed by inverting and then aliquoted into 10 petri dishes and placed in a 5% CO2 incubator for 10–14 days. Single-cell colonies were picked (5 µL) and placed in 96-well plates containing 10-DMEM in 200 µL. The cell supernatants were screened by ELISA with plates coated with Tn-BSA (4 µg/mL) when sufficient antibody was produced [15]. Tn-BSA was used in the selection protocol to avoid PS A1 interactions.
mIgM purification
Purification of mIgM antibodies was conducted according to literature procedure [35]. Cell culture supernatant was dialyzed against distilled water causing a precipitation of the mIgM antibody after 1 day at 4 °C. The resulting precipitate was centrifuged to remove water. The protein was dissolved in 10 mL of 1 × PBS buffer and this was followed by re-precipitation by adding 17.1 g of ammonium sulfate. The precipitate was then concentrated and purified using size exclusion chromatography (Sephacryl S-300). Collected fractions were individually checked for protein by UV monitoring at 280 nm. The resulting fractions containing mIgM antibody were pooled, sterile filtered, and stored at 4 °C.
Enzyme-linked immunosorbent assay
In-house generated monomeric BSA constructs [Tn-BSA, Thomsen–Friedenreich (TF)-BSA, etc.] were coated on Immulon® Microtiter™ 4 HBX 96 well plates using a carbonate buffer (pH 9.6) at a concentration of 4 µg/mL over night at 4 °C. The wells were then washed three times with 200 µL/well of washing buffer (1 × TBS with 0.1% TWEEN® 20) followed by incubation of 200 µL of a blocking buffer (3% BSA in 1 × TBS) for 1 h. Wells were washed three times with washing buffer, then 100 µL of a solution containing antibody (Kt-IgM-8 or Tn-218) in TBS was then incubated for 2 h at 37 °C. Wells were then washed three times with washing buffer. Rat anti-mouse IgM alkaline phosphatase conjugate (Southern Biotech, Catalog#:1139-04) was then diluted to 1:1000 in TBS and 100 µL of secondary antibody solution was then added to each well and incubated for 1 h at 37 °C. Wells were washed three times with washing buffer. A p-nitrophenylphosphate (PNPP) solution, in diethanolamine buffer (1 mg/mL), was then added to each well to a final volume of 100 µL. The PNPP solution was incubated for 30 min before measuring the OD (405 nm) using a BioTek PowerWave HT microplate spectrophotometer.
SDS-PAGE
Kt-IgM-8 was further purified using Pierce™ IgM purification kit (ThermoFischer Scientific, Cat#44897) per kit instructions. IgM samples [Kt-IgM-8 and mouse IgM, κ isotype control, clone: MM-30, (BioLegend Cat#401601)] were separately mixed 1:1 with a PAGE sample buffer containing 2-ME. 20 µL samples (0.5 mg/mL) were loaded into the stacking gel. A constant amperage of 30 mA was applied using a BIO-RAD mini-PROTEAN® II system for 25 min (supplementary Fig. 1). The gel was then stained using Coomassie blue, washed with water, and de-stained using 5% acetic acid.
Flow cytometry
Kt-IgM-8 was diluted to 30 µg/mL in FACS staining buffer (1 × PBS, 0.5% BSA, 0.1% sodium azide) and incubated with cell lines MCF-7 or human colorectal carcinoma 116 (HCT-116) (both at 2.0 × 106) for 30 min on ice and then washed three times. Cells were labeled with AlexaFluor® 647 (Southern Biotech Cat#1021-31) and data were acquired using Becton Dickinson (BD) FACSCalibur™ and analyzed with FlowJo™ software.
Complement-dependent cytotoxicity
2 × 104 MCF-7 cells were plated in a 96-well-plate overnight. The cells were exposed to 51Cr for 4 h, and then, the wells were washed with cell media. 100 µL of Kt-IgM-8 (30 µg/mL), or anti-Tn-PS A1 whole sera, or purified pIgGs from anti-Tn-PS A1 sera, or anti-PS A1 or anti-PBS whole sera was added to wells. The antibodies were incubated for 1 h at 37 °C in a 5% CO2 incubator. The cells were then washed with cell media and 10% complement was added to each well. 51Cr release was measured after 18 h using liquid scintillation to quantify 51Cr release, and % cytotoxicity was calculated using the following formula: (experimental − spontaneous)/(max − spontaneous) × 100. Spontaneous wells only received media.
SCID mice tumor implantation and adoptive transfer of immunotherapeutic
The SCIDs (Crl:SHO®-PrkdcscidHrhr) were surgically implanted with a 17β-estradiol 60-day release pellet (0.72 mg/pellet) (Innovative Research of America) behind the shoulders. After 2 days, 5 × 105 MCF-7 tumors cells were mixed with Geltrex® Matrix (1:1) at 4 °C and subcutaneously injected into the mice flanks (2 × per mouse). The mice tumors were measured with microcalipers three times a week using the equation [tumor volume mm3 = (length × width2)/2]. 4 days after tumor implantation, 200 µL of anti-Tn-PS A1 whole sera, or purified pIgGs from anti-Tn-PS A1 sera, or anti-PBS or Kt-IgM-8 (30 µg/mL) was i.p. injected once every week until the humane endpoint was reached. Each cohort consisted of four mice. Data were analyzed using GraphPad Prism and ANOVA was used for determining statistical significance.
Kt-IgM-8 sequencing of heavy and light chains
Hybridoma cell line secreting Kt-IgM-8 was sent to Vanderbilt University Antibody and Protein Resource core services for extraction of cDNA and sequencing of heavy and light chain Kt-IgM-8.
Glycan array
A glycan array was used to determine the binding specificities of Kt-IgM-8 and commercial anti-Tn IgM mAb Tn-218 (OriGene™ AM10039PU). Glycan array generation and antibody binding specificity were described as in Prendergast et al. [36], except experimental buffers were tailored to IgMs rather than IgGs. Briefly, Tn and 70 related glycans were synthesized using a one-pot three-enzyme chemoenzymatic approach, structures were confirmed by 1,2-diamino-4,5-methylenedioxybenzene (DMB)-HPLC, nuclear magnetic resonance spectroscopy (NMR) or mass spectrometry (MS) and printed and as described in the literature [36]. Synthesized glycans were diluted to a final concentration of 100 µM (300 mM phosphate buffer, pH 8.4) and printed in 4 replicates. Epoxy slides were blocked (0.1 M Tris, 0.05 M ethanol amine, pH 9.0) for 1 h at 50 °C. Slides were washed with distilled water and then blocked (PBS with 1% OVA) for 1 h. Blocking buffer was aspirated and anti-Tn antibody was added at 10 µg/mL concentration diluted in blocking buffer for 1 h. Slides were then washed twice with PBS with 0.1% TWEEN® 20, then once with PBS alone. Secondary antibody [cyanine 3 (Cy3) goat anti-mouse IgM, Jackson ImmunoResearch 115-165-075 or goat anti-mouse IgM AlexaFluor® 647 Southern Biotech 1021-31] was added at a final concentration of 5 µg/mL in PBS for 1 h. Slides were washed with PBS buffer then distilled water and finally air dried before reading on the GenePix 4000B (Molecular Devices, LLC). Fluorescence intensities were measured, and background noise was subtracted using the GenePix Pro software. Intensity of Tn (GalNAcα) was compared along with the remaining glycans on the array to determine binding selectivity. Binding was compared to 10 µg/mL mouse monoclonal IgM anti-Tn antibody Tn-218 (OriGene™ Cat#AM10039PU-S) and 10 µg/mL IgM isotype control (Clone MM-30, BioLegend Cat#401601). Positive and negative controls consisted of a commercial Tn-binding lectin [40 µg/mL Vicia Villosa Lectin (VVL)-biotin, Vector Laboratories Cat#B-1235; and 5 µg/mL streptavadin-Cy3 Jackson ImmunoResearch Cat#016-160-084] and IgM secondary only, respectively.
Results and discussion
PS A1 was chosen as an immune stimulant, because it is a ZPS capable of inducing T-cell immune responses for antibodies avoiding unwanted Fab donor–acceptor interactions other than those of carbohydrates. After immunizing mice, spleen cells were homogenized to single cell suspensions and fused with the myeloma cell line Sp2/0-Ag14 [37]. The resulting hybridoma cell culture supernatants were screened to bind with the Tn antigen that was conjugated to BSA. The hybridoma cell supernatant that demonstrated the best binding events to Tn-BSA was chosen for scale-up procedures for in vitro and in vivo studies. The optimal working concentration of Kt-IgM-8 (Fig. 2a, b) was determined by serially diluting Kt-IgM-8 (60–0.01 µg/mL) on 96-well plates coated with 4 µg/mL Tn-BSA. Optimal binding in the titration of the antibody to Tn was observed at 0.3 µg/mL with an OD of greater than 0.2. For a mIgM antibody, binding at such low concentrations to a carbohydrate rivals that of an mIgG antibody and indicates high avidity due to the pentavalent nature of the mIgM itself [38]. A commercial Tn-binding mIgM antibody, Tn-218, was then used for comparison and concentrations for antibody binding of the Tn antigen on ELISA (Fig. 2c) were tested. Kt-IgM-8 and Tn-218 were screened in parallel at an initial concentration of 30 µg/mL and serially diluted to a final concentration of 0.23 µg/mL. Surprisingly, the commercial Tn-218 only minimally recognized the Tn sugar when Tn-BSA was used as the coating construct most likely due to the non-multimeric presentation of Tn in Tn-BSA. In contrast, Kt-IgM-8 showed enhanced binding towards Tn when the same coating construct was used (Fig. 2c). To expand on the specificity of Kt-IgM-8, a panel of TACA-related constructs was prepared in-house or purchased, which all displayed various Tn-like and Tn antigens (α/β-Tn-Thr-BSA, α-Tn-BSA, α-TF-BSA, blood group A, and blood group B) for screening by ELISA (Fig. 2c). From this study, we observed that Kt-IgM-8 had no discernible binding preference between α or β containing Tn-Thr glycosides [noted in red and green (Fig. 2d)]; however, there was a notable decreased binding event when α-TF-BSA construct (TF = β-d-Galp-(1,3)-α-d-GalpNAc) was used [noted in purple (Fig. 2d)]. In addition, Kt-IgM-8 did not bind to PS A1 (used as control) or BSA (used as a blocking agent) on the ELISA plates (data not shown). Kt-IgM-8 did not recognize blood groups A or B below 30 µg/mL (OD ≤ 0.2), but did partially bind blood groups A and B at increased concentration [60 µg/mL (OD ≥ 0.2)]. This result suggested that Kt-IgM-8 will not likely promote hemolytic activity due to structure similarities of α-GalpNAc between Tn and blood groups A and B. Overall, Tn-PS A1 immunization produced an antibody with an enriched Tn antigen reactivity and it exceeded the monovalent Tn-binding events observed from other mAbs produced from protein sources such as commercially available monoclonal Tn-218 (Fig. 2c).

Characterization and titrations of Kt-IgM-8 and Tn-218 in ELISA. a OD values of Kt-IgM-8 binding to Tn-BSA from concentrations 60–0.01 µg/mL. b OD values of Kt-IgM-8 binding to Tn-BSA from concentrations 0.8–0.01 µg/mL. c Kt-IgM-8 and Tn-218 binding to Tn-BSA from concentrations 30–0.01 µg/mL. d Cross reactivity of Kt-IgM-8 with other Tn-like or GalNAc-containing glycans from concentrations 60–0.47 µg/mL
Our next step in profiling Kt-IgM-8 was to determine if the antibody could bind to cancer cells known to express the Tn antigen on the cell surface by FACS. MCF-7 and HCT-116 were chosen as both have cell surface Tn antigen [39, 40] and they represent two of the most common forms of cancers [41]. This in vitro experiment suggests feasibility for additional immunotherapy in in vivo models. We chose mouse anti-IgM AlexaFluor® 647 as the fluorescent secondary antibody to detect our primary mIgM (Kt-IgM-8) antibody in FACS. Kt-IgM-8 demonstrated the ability to bind both MCF-7 and HCT-116 tumor cells at 30 µg/mL (Fig. 3) with a shift in fluorescence of 49% in both cell lines. Collectively, the presence and recognition of Tn by Kt-IgM-8, as confirmed by FACS (Fig. 3), further validate Tn selectivity that was initially demonstrated by ELISA.

Flow cytometry of Kt-IgM-8 binding to Tn expressing cancer cell lines. a MCF-7 and b HCT-116
Antibody function was assessed using a 51Cr CDC assay with MCF-7s. Kt-IgM-8, anti-Tn-PS A1 whole sera, purified pIgGs from anti-Tn-PS A1 sera, anti-PS A1 sera, and control sera from PBS immunizations were used in comparison with CDC activity (Fig. 4). Both the anti-Tn-PS A1 whole sera and purified pIgGs from anti-Tn-PS A1 sera were used as cytotoxicity controls. The purified pIgGs from anti-Tn-PS A1 sera were essential in determining how effective IgGs from immunizations could be at initiating CDC in the absence of pIgMs. We observed that Kt-IgM-8 had the greatest CDC activity out of the tested antibodies at ~ 30% cytotoxicity and this was statistically significant compared to both anti-Tn-PS A1 sera (15%, p < 0.005) and purified pIgGs from anti-Tn-PS A1 sera (8%, p < 0.005). CDC activity was absent from anti-serums of PS A1 and PBS control immunized mice. From an immunotherapeutic perspective, Kt-IgM-8 can initiate CDC at a greater rate than what was determined from other immunizations as a correlation to the overall concentration of antibody used. Based on the observed data, we further hypothesized that a Tn-selective mIgM antibody would provide protection in in vivo tumor mouse models through complement-mediated cytotoxicity.

CDC activity of Kt-IgM-8 on MCF-7 cells. Data are illustrated as mean ± sem. **p < 0.005, ***p < 0.0005; two tailed Student’s t test
To test our hypothesis, we turned our attention to SCID mice which lack functional immune responses (both B and T lymphocytes), but maintain an intact complement protein system, allowing for implantation and study of human tumors for CDC models [42]. We focused on MCF-7s as a model system for studying breast cancer in SCID mice. MCF-7 tumor growth in SCID mice was determined by measuring tumor volume and immunotherapeutic efficacy was assessed by comparing tumor volume in the control mice (PBS). Figure 5 presents four different treatments: anti-PBS as control, Kt-IgM-8 (Fig. 5a), anti-Tn-PS A1 whole sera (Fig. 5b), and purified pIgGs from anti-Tn-PS A1 sera (Fig. 5c). The humane endpoint of the experiment was determined when tumor volume approached 400 mm3. The control mice, treated with PBS (vehicle), should not convey any immunogenicity to protect against the tumors and have thus been used to determine the efficacy of each therapeutic. The anti-Tn-PS A1 whole sera provided the greatest protection against tumor growth at 52% compared to PBS (Fig. 5d). This might be because the anti-Tn-PS A1 whole sera, which contained both pIgGs and pIgMs, was more effective at recruiting complement in a CDC (Fig. 4) mode of action. The purified pIgGs from anti-Tn-PS A1 sera did not show a statistically significant reduction in tumor growth when compared to the PBS control mice. A large portion of pIgGs were shown to recognize PS A1, diminishing overall specificity towards Tn and consequently tumor reduction (data not shown). Kt-IgM-8 also demonstrated protection against tumors at a 39% compared to PBS (Fig. 5d), which defines the effectiveness of the treatment and their role in minimizing tumor growth.

Kt-IgM-8 displays tumor volume (mm3) reduction of MCF-7 tumors in SCID mice for 39 days. a Kt-IgM-8 treatment of MCF-7 tumor growth in comparison with PBS control mice. b Anti-Tn-PS A1 whole sera in comparison with PBS mice. c Purified pIgGs from anti-Tn-PS A1 sera in comparison with PBS mice. d Tumor volume at day 39. Data are illustrated as mean ± sem. **p < 0.005, ***p < 0.001; ANOVA
The glycan array results signify preferential selectivity to the Tn antigen. Kt-IgM-8 was visualized to bind to the printed, monovalent Tn antigen (spot 47) more so than other glycans (for full list of array glycans see supplementary Table 1 and Prendergast et al.) [36]. However, Kt-IgM-8 does also bind a limited number of sialylated glycans of the Neu5Gc type (1.3–2.6-fold lower than Tn binding, see Fig. 6) more often than the Neu5Ac type. Neu5Gc sialic acids are not normally expressed in humans, but can be consumed through the diet, and their presence is enriched in tumorous and other diseased tissues [43]. Two secondary antibodies were used for this experiment (Cy3 and A647) in which glycan-binding trends were consistent, but differences in fluorescence intensity were observed. When compared to the commercially available anti-Tn IgM, Tn-218, Kt-IgM-8 demonstrated superior binding to Tn on the glycan array. Tn-218 failed to bind the monovalent Tn antigen at similar or increased concentrations (5, 12.5, 25, and 50 µg/mL, data not shown). These results are promising as similar anti-Tn IgMs have struggled to preferentially bind monovalent Tn as presented on glycan array surfaces.

Bar chart representation of glycan array results. Tn is number 47 on the array and for a full list of glycan structures see SI Table 1 and Prendergast et al. [39]
The variable regions of the heavy and light chains of Kt-IgM-8 were sequenced as noted in supplementary Figs. 2 and 3. The sequencing allows bioinformatic identification of variable regions that bind specifically to carbohydrate-based antigens, which can give further insight into immune recognition. In addition, sequencing of the variable regions can be used for humanizing the Kt-IgM-8 by insertion of the heavy and light chains into human antibodies which can be used as diagnostic tools or immunotherapeutics.
Conclusions
In conclusion, the zwitterionic nature of PS A1 evokes a natural CD4+ immune response, which can assist in the production of unique anti-glycan antibodies. To validate our approach to immunotherapy, we adapted PS A1 to accommodate the Tn antigen for cancer intervention. The rationale for using an entirely carbohydrate immunogen [Tn-PS A1 (1)] was to focus the antibody recognition on glycosides to generate antibodies that have no binding preference to peptides or lipids. The pentavalent binding nature of mIgM Kt-IgM-8 in combination with our observations noted herein suggest that IgM antibodies may confer an advantage for this particular glycan antigen. Tn can be associated with a wide variety of peptide carriers and an immunotherapeutic that recognizes glycosides independent of this carrier can be a beneficial feature. Producing a mAb for glycosides with our entirely carbohydrate immunogen can lead to preferential Tn antigen binding, avoiding cross reactivity with peptides that are naturally occurring.
IgG antibodies may not always be the preferred therapeutic choice as they relate to sugar antigens, and IgM antibodies may be a viable alternative to elicit CDC responses. Kt-IgM-8 also represents a biological tool that has demonstrated in vitro complement activity and in vivo inhibition of tumor progression in an SCID mice model compared to PBS (vehicle) controls. Few commercially available anti-Tn antibodies have been used for generating in vivo data (MLS 128, GOD3-2C4, and KM3413) all of which are of the IgG type [44,45,46]. For example, GOD3-2C4 IgG was the only anti-Tn antibody to be used in an adoptive transfer in vivo model in SCID mice, but in vitro activity was only assessed by antibody-dependent cellular cytotoxicity (ADCC), while CDC was not examined [44]. Kt-IgM-8 also represents a select few IgM antibodies that utilizes CDC to be used in passive immunotherapy preferentially targeting the carbohydrate cancer antigen Tn. However, at the current stage of development, Kt-IgM-8 is unlikely to be a useful therapeutic agent due to its marginal carbohydrate specificity.
Abbreviations
- 10-DMEM:
-
10% FCS-DMEM
- 20-DMEM:
-
20% FCS-DMEM
- ATCC:
-
American-type culture collection
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- BD:
-
Becton Dickinson
- CDC:
-
Complement-dependent cytotoxicity
- Cy3:
-
Cyanine 3
- DMB:
-
1,2-Diamino-4,5-methylenedioxybenzene
- FDA:
-
Food and Drug Administration
- GD2:
-
Ganglioside disialic acid 2
- HCT-116:
-
Human colorectal carcinoma
- KLH:
-
Keyhole limpet hemocyanin
- MCF-7:
-
Michigan Cancer Foundation breast cell line 7
- mIgG:
-
Monoclonal IgG
- mIgM:
-
Monoclonal IgM
- MS:
-
Mass spectrometry
- NCI:
-
National Cancer Institute
- NCTC:
-
National collection of type cultures
- NMR:
-
Nuclear magnetic resonance spectroscopy
- pIgG:
-
Polyclonal IgG
- pIgM:
-
Polyclonal IgM
- PNPP:
-
Para-nitrophenylphosphate
- PS A1:
-
Polysaccharide A1
- STn:
-
Sialyl Thomsen-nouveau
- TACA:
-
Tumor-associated carbohydrate antigen
- TF:
-
Thomsen–Friedenreich
- Tn:
-
Thomsen-nouveau
- VVL:
-
Vicia villosa lectin
- ZPS:
-
Zwitterionic polysaccharide
References
Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM (2009) The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 15(17):5323–5337. https://doi.org/10.1158/1078-0432.ccr-09-0737
O’Cearbhaill RE, Ragupathi G, Zhu J, Wan Q, Mironov S, Yang G, Spassova MK, Iasonos A, Kravetz S, Ouerfelli O, Spriggs DR, Danishefsky SJ, Sabbatini PJ (2016) A phase I study of unimolecular pentavalent (Globo-H-GM2-STn-TF-Tn) immunization of patients with epithelial ovarian, fallopian tube, or peritoneal cancer in first remission. Cancers (Basel). https://doi.org/10.3390/cancers8040046
Laubreton D, Bay S, Sedlik C, Artaud C, Ganneau C, Deriaud E, Viel S, Puaux AL, Amigorena S, Gerard C, Lo-Man R, Leclerc C (2016) The fully synthetic MAG-Tn3 therapeutic vaccine containing the tetanus toxoid-derived TT830-844 universal epitope provides anti-tumor immunity. Cancer Immunol Immunother 65(3):315–325. https://doi.org/10.1007/s00262-016-1802-0
Scheid E, Major P, Bergeron A, Finn OJ, Salter RD, Eady R, Yassine-Diab B, Favre D, Peretz Y, Landry C, Hotte S, Mukherjee SD, Dekaban GA, Fink C, Foster PJ, Gaudet J, Gariepy J, Sekaly RP, Lacombe L, Fradet Y, Foley R (2016) Tn-MUC1 DC vaccination of rhesus macaques and a phase I/II trial in patients with nonmetastatic castrate-resistant prostate cancer. Cancer Immunol Res 4(10):881–892. https://doi.org/10.1158/2326-6066.cir-15-0189
Gilewski T, Ragupathi G, Bhuta S, Williams LJ, Musselli C, Zhang XF, Bornmann WG, Spassova M, Bencsath KP, Panageas KS, Chin J, Hudis CA, Norton L, Houghton AN, Livingston PO, Danishefsky SJ (2001) Immunization of metastatic breast cancer patients with a fully synthetic Globo H conjugate: a phase I trial. Proc Natl Acad Sci USA 98(6):3270–3275. https://doi.org/10.1073/pnas.051626298
Son HY, Apostolopoulos V, Kim CW (2016) T/Tn immunotherapy avoiding immune deviation. Int J Immunopathol Pharmacol 29(4):812–817. https://doi.org/10.1177/0394632016674018
Dhillon S (2015) Dinutuximab: first global approval. Drugs 75(8):923–927. https://doi.org/10.1007/s40265-015-0399-5
Aggarwal RS (2014) What’s fueling the biotech engine—2012 to 2013. Nat Biotechnol 32(1):32–39. https://doi.org/10.1038/nbt.2794
Weiner GJ (2015) Building better monoclonal antibody-based therapeutics. Nat Rev Cancer 15(6):361–370. https://doi.org/10.1038/nrc3930
Buskas T, Li Y, Boons GJ (2004) The immunogenicity of the tumor-associated antigen Lewis(Y) may be suppressed by a bifunctional cross-linker required for coupling to a carrier protein. Chem Eur J 10(14):3517–3524. https://doi.org/10.1002/chem.200400074
Kudryashov V, Glunz PW, Williams LJ, Hintermann S, Danishefsky SJ, Lloyd KO (2001) Toward optimized carbohydrate-based anticancer vaccines: Epitope clustering, carrier structure, and adjuvant all influence antibody responses to Lewis(Y) conjugates in mice. Proc Natl Acad Sci USA 98(6):3264–3269. https://doi.org/10.1073/pnas.051623598
Kagan E, Ragupathi G, Yi SS, Reis CA, Gildersleeve J, Kahne D, Clausen H, Danishefsky SJ, Livingston PO (2005) Comparison of antigen constructs and carrier molecules for augmenting the immunogenicity of the monosaccharide epithelial cancer antigen Tn. Cancer Immunol Immunother 54(5):424–430. https://doi.org/10.1007/s00262-004-0584-y
Ibrahim NK, Murray JL (2003) Clinical development of the STn-KLH vaccine (THERATOPE®). Clin Breast Cancer 3(Suppl 4):S139–S143. https://doi.org/10.3816/CBC.2003.s.003
Holmberg LA, Sandmaier BM (2001) Theratope vaccine (STn-KLH). Expert Opin Biol Ther 1(5):881–891. https://doi.org/10.1517/14712598.1.5.881
De Silva RA, Wang Q, Chidley T, Appulage DK, Andreana PR (2009) Immunological response from an entirely carbohydrate antigen: design of synthetic vaccines based on Tn-PS A1 conjugates. J Am Chem Soc 131(28):9622–9623. https://doi.org/10.1021/ja902607a
Kalia J, Raines RT (2008) Hydrolytic stability of hydrazones and oximes. Angew Chem Int Ed Engl 47(39):7523–7526. https://doi.org/10.1002/anie.200802651
De Silva RA, Appulage DK, Pietraszkiewicz H, Bobbitt KR, Media J, Shaw J, Valeriote FA, Andreana PR (2012) The entirely carbohydrate immunogen Tn-PS A1 induces a cancer cell selective immune response and cytokine IL-17. Cancer Immunol Immunother 61(4):581–585. https://doi.org/10.1007/s00262-012-1205-9
Shi M, Kleski KA, Trabbic KR, Bourgault JP, Andreana PR (2016) Sialyl-Tn polysaccharide A1 as an entirely carbohydrate immunogen: synthesis and immunological evaluation. J Am Chem Soc 138(43):14264–14272. https://doi.org/10.1021/jacs.6b05675
Irie RF, Ollila DW, O’Day S, Morton DL (2004) Phase I pilot clinical trial of human IgM monoclonal antibody to ganglioside GM3 in patients with metastatic melanoma. Cancer Immunol Immunother 53(2):110–117. https://doi.org/10.1007/s00262-003-0436-1
Liedtke M, Twist CJ, Medeiros BC, Gotlib JR, Berube C, Bieber MM, Bhat NM, Teng NN, Coutre SE (2012) Phase I trial of a novel human monoclonal antibody mAb216 in patients with relapsed or refractory B-cell acute lymphoblastic leukemia. Haematologica 97(1):30–37. https://doi.org/10.3324/haematol.2011.045997
Melis JP, Strumane K, Ruuls SR, Beurskens FJ, Schuurman J, Parren PW (2015) Complement in therapy and disease: regulating the complement system with antibody-based therapeutics. Mol Immunol 67(2 Pt A):117–130. https://doi.org/10.1016/j.molimm.2015.01.028
Brandlein S, Pohle T, Ruoff N, Wozniak E, Muller-Hermelink HK, Vollmers HP (2003) Natural IgM antibodies and immunosurveillance mechanisms against epithelial cancer cells in humans. Cancer Res 63(22):7995–8005
Imai M, Landen C, Ohta R, Cheung NK, Tomlinson S (2005) Complement-mediated mechanisms in anti-GD2 monoclonal antibody therapy of murine metastatic cancer. Cancer Res 65(22):10562–10568. https://doi.org/10.1158/0008-5472.can-05-1894
Czajkowsky DM, Shao Z (2009) The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. Proc Natl Acad Sci USA 106(35):14960–14965. https://doi.org/10.1073/pnas.0903805106
Ehrenstein MR, Notley CA (2010) The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol 10(11):778–786. https://doi.org/10.1038/nri2849
Manimala JC, Roach TA, Li Z, Gildersleeve JC (2007) High-throughput carbohydrate microarray profiling of 27 antibodies demonstrates widespread specificity problems. Glycobiology 17(8):17c–23c. https://doi.org/10.1093/glycob/cwm047
Karsten U, Diotel C, Klich G, Paulsen H, Goletz S, Muller S, Hanisch FG (1998) Enhanced binding of antibodies to the DTR motif of MUC1 tandem repeat peptide is mediated by site-specific glycosylation. Cancer Res 58(12):2541–2549
Karsten U, Serttas N, Paulsen H, Danielczyk A, Goletz S (2004) Binding patterns of DTR-specific antibodies reveal a glycosylation-conditioned tumor-specific epitope of the epithelial mucin (MUC1). Glycobiology 14(8):681–692. https://doi.org/10.1093/glycob/cwh090
Ohyabu N, Kakiya K, Yokoi Y, Hinou H, Nishimura S (2016) Convergent solid-phase synthesis of macromolecular MUC1 models truly mimicking serum glycoprotein biomarkers of interstitial lung diseases. J Am Chem Soc 138(27):8392–8395. https://doi.org/10.1021/jacs.6b04973
Rangappa S, Artigas G, Miyoshi R, Yokoi Y, Hayakawa S, Garcia-Martin F, Hinou H, Nishimura S-I (2016) Effects of the multiple O-glycosylation states on antibody recognition of the immunodominant motif in MUC1 extracellular tandem repeats. MedChemComm 7(6):1102–1122. https://doi.org/10.1039/C6MD00100A
Li Q, Anver MR, Butcher DO, Gildersleeve JC (2009) Resolving conflicting data on expression of the Tn antigen and implications for clinical trials with cancer vaccines. Mol Cancer Ther 8(4):971–979. https://doi.org/10.1158/1535-7163.mct-08-0934
Oyelaran O, Li Q, Farnsworth D, Gildersleeve JC (2009) Microarrays with varying carbohydrate density reveal distinct subpopulations of serum antibodies. J Proteome Res 8(7):3529–3538. https://doi.org/10.1021/pr9002245
Jegerlehner A, Wiesel M, Dietmeier K, Zabel F, Gatto D, Saudan P, Bachmann MF (2010) Carrier induced epitopic suppression of antibody responses induced by virus-like particles is a dynamic phenomenon caused by carrier-specific antibodies. Vaccine 28(33):5503–5512. https://doi.org/10.1016/j.vaccine.2010.02.103
Schutze MP, Leclerc C, Jolivet M, Audibert F, Chedid L (1985) Carrier-induced epitopic suppression, a major issue for future synthetic vaccines. J Immunol 135(4):2319–2322
Andrew SM, Titus JA, Coico R, Amin A (2001) Purification of immunoglobulin M and immunoglobulin D. Curr Protoc Immunol. https://doi.org/10.1002/0471142735.im0209s21 (Chap. 2:Unit 2.9)
Prendergast JM, Galvao da Silva AP, Eavarone DA, Ghaderi D, Zhang M, Brady D, Wicks J, DeSander J, Behrens J, Rueda BR (2017) Novel anti-sialyl-Tn monoclonal antibodies and antibody-drug conjugates demonstrate tumor specificity and anti-tumor activity. MAbs 9(4):615–627. https://doi.org/10.1080/19420862.2017.1290752
Kudryashov V, Ragupathi G, Kim IJ, Breimer ME, Danishefsky SJ, Livingston PO, Lloyd KO (1998) Characterization of a mouse monoclonal IgG3 antibody to the tumor-associated Globo H structure produced by immunization with a synthetic glycoconjugate. Glycoconj J 15(3):243–249. https://doi.org/10.1023/A:1006992911709
Heimburg-Molinaro J, Rittenhouse-Olson K (2009) Development and characterization of antibodies to carbohydrate antigens. Methods Mol Biol 534:341–357. https://doi.org/10.1007/978-1-59745-022-5_24
Freire T, Bay S, von Mensdorff-Pouilly S, Osinaga E (2005) Molecular basis of incomplete O-glycan synthesis in MCF-7 breast cancer cells: putative role of MUC6 in Tn antigen expression. Cancer Res 65(17):7880–7887. https://doi.org/10.1158/0008-5472.can-04-3746
Oura F, Yajima Y, Nakata M, Taniue K, Akiyama T, Nakada H, Yamamoto K, Fujita-Yamaguchi Y (2015) Susceptibility to proteases of anti-Tn-antigen MLS128 binding glycoproteins expressed in human colon cancer cells. Biosci Trends 9(1):49–55. https://doi.org/10.5582/bst.2014.01127
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63(1):11–30. https://doi.org/10.3322/caac.21166
Koo GC, Hasan A, O’Reilly RJ (2009) Use of humanized severe combined immunodeficient mice for human vaccine development. Expert Rev Vaccines 8(1):113–120. https://doi.org/10.1586/14760584.8.1.113
Samraj AN, Pearce OM, Laubli H, Crittenden AN, Bergfeld AK, Banda K, Gregg CJ, Bingman AE, Secrest P, Diaz SL, Varki NM, Varki A (2015) A red meat-derived glycan promotes inflammation and cancer progression. Proc Natl Acad Sci USA 112(2):542–547. https://doi.org/10.1073/pnas.1417508112
Welinder C, Baldetorp B, Borrebaeck C, Fredlund BM, Jansson B (2011) A new murine IgG1 anti-Tn monoclonal antibody with in vivo anti-tumor activity. Glycobiology 21(8):1097–1107. https://doi.org/10.1093/glycob/cwr048
Ando H, Matsushita T, Wakitani M, Sato T, Kodama-Nishida S, Shibata K, Shitara K, Ohta S (2008) Mouse-human chimeric anti-Tn IgG1 induced anti-tumor activity against Jurkat cells in vitro and in vivo. Biol Pharm Bull 31(9):1739–1744. https://doi.org/10.1248/bpb.31.1739
Nakada H, Inoue M, Numata Y, Tanaka N, Funakoshi I, Fukui S, Mellors A, Yamashina I (1993) Epitopic structure of Tn glycophorin a for an anti-Tn antibody (MLS 128). Proc Natl Acad Sci USA 90(6):2495–2499. https://doi.org/10.1073/pnas.90.6.2495
Acknowledgements
We are grateful to Fred Valeriote, Joe Media, and Halina Pietrazkiewicz (Henry Ford Health System) for donating human tumor cell lines as well as insightful discussions regarding SCID mice models. In addition, we would like to thank Katherine Goans (DLAR, University of Toledo) for assistance with surgical xenograft procedures on SCID mice.
Funding
The research was funded by the National Institutes of Health/National Cancer Institute under Grant 1R01 CA156661 and in part by the National Cancer Institute of the National Institutes of Health Small Business Innovation Research under Grants 1R43CA186326-01A1, HHSN261200700063C and HHSN261200900034C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
KRT contributed to the majority of work including preparation of Tn-PS A1 constructs, murine immunizations, production and isolation of Kt-IgM-8, ELISA, FACS, CDC assay, and SCID mice studies. KAK expressed and purified Kt-IgM-8 for SDS-PAGE and prepared samples for glycan arrays. MS synthesized the Tn antigen used for conjugation. J-PB assisted in the SCID mice studies. JMP conducted glycan array experiments. DTD conducted glycan array experiments. PRA is the principle investigator and director of the research.
Corresponding author
Ethics declarations
Conflict of interest
Kevin R. Trabbic, Kristopher A. Kleski, Mengchao Shi, Jean-Paul Bourgault, and Peter R. Andreana declare that they have no conflict of interest. The glycan array described is owned by and/or licensed to Siamab Therapeutics, Inc. in which Jillian M. Prendergast and Daniel T. Dransfield have a business and/or financial interest.
Ethical approval and ethical standards
C57BL/6 male mice (6 weeks) were obtained from Jackson Laboratories and maintained by the DLAR at the University of Toledo. All animal protocols were approved and performed in compliance with the relevant laws and institutional guidelines set forth by the IACUC of the University of Toledo (Protocol 107956). SHO® 4-week-old female mice (Crl:SHO®-PrkdcscidHrhr) were obtained from Charles River Laboratories and maintained by the DLAR at the University of Toledo. All animal protocols were approved and performed in compliance with the relevant laws and institutional guidelines set forth by the IACUC of the University of Toledo (Protocol Number 108420). The Sp2/0-Ag14 (ATCC CRL 1581) cell line was purchased from the American-Type Culture Collection. The MCF-7 (ATCC HTB-22) cell line and the HCT-116 (ATCC CCL-247) cell line was from Henry Ford Health Systems.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Trabbic, K.R., Kleski, K.A., Shi, M. et al. Production of a mouse monoclonal IgM antibody that targets the carbohydrate Thomsen-nouveau cancer antigen resulting in in vivo and in vitro tumor killing. Cancer Immunol Immunother 67, 1437–1447 (2018). https://doi.org/10.1007/s00262-018-2206-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-018-2206-0