
Abstract
The ability of some tumors to exclude effector T cells represents a major challenge to immunotherapy. T cell exclusion is particularly evident in pancreatic ductal adenocarcinoma (PDAC), a disease where blockade of the immune checkpoint molecule CTLA-4 has not produced significant clinical activity. In PDAC, effector T cells are often scarce within tumor tissue and confined to peritumoral lymph nodes and lymphoid aggregates. We hypothesized that CTLA-4 blockade, despite a lack of clinical efficacy seen thus far in PDAC, might still alter T cell immunobiology, which would have therapeutic implications. Using clinically relevant genetic models of PDAC, we found that regulatory T cells (Tregs), which constitutively express CTLA-4, accumulate early during tumor development but are largely confined to peritumoral lymph nodes during disease progression. Tregs were observed to regulate CD4+, but not CD8+, T cell infiltration into tumors through a CTLA-4/CD80 dependent mechanism. Disrupting CTLA-4 interaction with CD80 was sufficient to induce CD4 T cell infiltration into tumors. These data have important implications for T cell immunotherapy in PDAC and demonstrate a novel role for CTLA-4/CD80 interactions in regulating T cell exclusion. In addition, our findings suggest distinct mechanisms govern CD4+ and CD8+ T cell infiltration in PDAC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite promising clinical activity seen with immunotherapy across a wide range of solid and hematologic malignancies, pancreatic ductal adenocarcinoma (PDAC) has demonstrated striking resistance to T cell immunotherapies, including adoptive T cell therapy [1, 2], vaccines [3, 4], and checkpoint inhibitors [5, 6]. This has been attributed to poor antigenicity and decreased immunogenicity of malignant cells as well as a strong immunosuppressive microenvironment [7]. Thus, understanding mechanisms of this biology will be critical for effectively applying immunotherapy to PDAC.
Recent success with overcoming T cell tolerance in cancer has involved the use of antibodies that target checkpoint molecules that regulate T cell priming and activation [8]. Cytotoxic lymphocyte-associated antigen-4 (CTLA-4) is an immune checkpoint molecule expressed on regulatory T cells (Tregs) as well as recently activated conventional T cells [9]. CTLA-4 can attenuate T cell responses by competing for ligands, including CD80 and CD86, which provide co-stimulatory signals to T cells via CD28. While targeting CTLA-4 with a human anti-CTLA4 antibody in patients with metastatic melanoma has produced durable tumor regressions in a subset of patients [10], this strategy has not demonstrated significant activity in patients with advanced PDAC [5]. Moreover, treatment combinations using anti-CTLA-4 antibodies with gemcitabine chemotherapy [11] or an allogeneic irradiated tumor cell vaccine (GVAX) [3] have shown encouraging, yet not statistically significant results in PDAC. With little benefit achieved so far with applying CTLA-4 antibodies to PDAC, it remains unclear whether this immune checkpoint molecule is a relevant therapeutic target in this malignancy.
Here, we investigated CTLA-4 and its impact in regulating T cell exclusion in PDAC using clinically relevant mouse models of this disease. Our findings show that blocking antibodies directed against CTLA-4 or its cognate receptor, CD80, can stimulate CD4+ T cell infiltration into spontaneously arising tumors. However, blocking CTLA-4/CD80 interactions is ineffective in directing CD8+ T cell infiltration into tumors and suggests distinct mechanisms regulating CD4+ and CD8+ T cell exclusion in PDAC. In addition, our findings suggest that strategies designed to overcome CD8+ T cell exclusion will likely be necessary to realize the potential of CTLA-4 blocking antibodies in PDAC.
Materials and methods
Mouse studies
Kras G12D/+;Trp53 R172H/+;Pdx-1Cre (KPC) mice and Mist1CreERT2;LSLKras G12D/+ (KCiMist1) mice were described previously [12, 13]. Pancreatic tumors were identified in KPC mice by abdominal palpation and confirmed by ultrasonography [14]. Tumor-bearing KPC mice were enrolled into treatment studies to receive antibodies against CTLA-4 (9H10, BioXcell), CD25 (PC61.5.3, BioXcell), or CD80 (16-10A1, BioXcell) versus control. Anti-CTLA-4 and anti-CD80 antibodies were administered bi-weekly, whereas anti-CD25 was administered once. Mice were euthanized 14 days after beginning treatment with tissues collected for subsequent analyses. All animal protocols were reviewed and approved by the Institute of Animal Care and Use Committee.
Treg quantification from Human Protein Atlas
Primary tumor tissue biopsies stained for Foxp3 were analyzed on the Human Protein Atlas public database [15, 16]. Positively stained cells were quantified in biopsies of 8–12 patients per cancer type.
Histology, immunohistochemistry, and immunofluorescence analysis
Immunohistochemistry on frozen tissue sections or formalin-fixed paraffin embedded tissues was performed as previously described [17]. Immunohistochemical staining of paraffin tissues was performed using the Ventana Discovery Ultra automated staining system. For frozen tissue staining, primary antibodies included anti-CD3 (Biorad), anti-CD4 (BioXcell), anti-CD8 (BioXcell), anti-Foxp3 (eBioscience), and anti-CD11c (Biolegend). Staining was visualized using a biotinylated anti-rat secondary (BD Biosciences) followed by avidin–biotin-HRP complex amplification (Vectastain) with DAB detection. For paraffin tissue staining, primary antibodies included anti-CD4 (Abcam). For two-color immunohistochemistry to detect CD11c and Foxp3, staining was performed sequentially with detection of CD11c using DAB plus nickel (black) and detection of Foxp3 using DAB alone (brown). Brightfield images were acquired on a BX43 upright microscope (Olympus).
Flow cytometry
Flow cytometry was performed on a FACSCanto II cytometer (BD Biosciences) using antibodies against Foxp3 (eBioscience, FJK-16 s), CD4 (BD Biosciences, RM4-5), and CTLA-4 (eBioscience, UC10-4B9). LIVE/DEAD Fixable Dead Cell Stain Kit (Life Technologies) was used to exclude dead cells. Cells were isolated from the pancreas by digestion with collagenase P. Collected data were analyzed using FlowJo software (Tree Star).
Statistical analysis
Significance was determined by Student’s t test or ANOVA with Tukey’s post hoc test for multiple comparisons.
Results
Tregs infiltrate early during pancreatic tumorigenesis but are largely confined to peritumoral lymph nodes during tumor progression
CTLA-4 is constitutively expressed by Foxp3+ regulatory T cells (Tregs) and in human cancers, Foxp3+ Tregs are reported at variable frequencies. Using the Human Protein Atlas [15, 16], we first quantified expression of Foxp3, a bona fide marker for Tregs [18, 19], in primary tumors of several solid cancers using immunohistochemistry. We found that Foxp3+ Tregs were detected in human pancreatic cancer at a frequency comparable to colorectal and lung cancers and higher than that seen in melanoma and prostate cancer (Fig. 1a, b).

Tregs accumulate in pancreatic cancer but predominately reside in peritumoral lymph nodes. a Foxp3+ cells were quantified in primary tumor specimens available from Human Protein Atlas database. Data points represent values from individual patients, with 8–12 patients per cancer type. b Shown are representative images of Foxp3 staining in solid tumors available from Human Protein Atlas. Scale bar is 100 μm. c The frequency of Foxp3+ cells in the blood (n = 16), spleen (n = 13) and whole pancreas (n = 14), which includes peripancreatic lymph nodes and normal adjacent pancreas tissue, of KPC mice was determined by flow cytometry. Shown is: d quantification per mm2 and e representative images of immunohistochemistry to detect Foxp3+ cells in pancreatic intraepithelial neoplasia (PanIN) (n = 7), pancreatic ductal adenocarcinoma (PDAC) (n = 10), peritumoral lymph nodes (n = 6), and control lymph nodes (n = 4) from KPC mice. Statistical significance was determined by one-way ANOVA and Tukey’s post hoc test. *p < 0.05; ***p < 0.001
To investigate the significance of Tregs in regulating the immune microenvironment in PDAC, we examined for Foxp3+ Tregs in two genetic mouse models of pancreatic ductal adenocarcinoma (PDAC). In the Mist1CreERT2;LSL-Kras G12D/+ (KCiMist1) mouse model of PDAC, we detected an increase in Foxp3+ Tregs in the pancreas compared with Mist1CreERT2 (CiMist1) control mice. The increase in Tregs in KCiMist1 mice was even more marked after induction of chronic pancreatitis using cerulein (Supplementary Fig. 1). In this model, cerulein-induced chronic inflammation drives carcinogenesis and the development of invasive PDAC [12]. While an increased frequency of Tregs was also observed after cerulein treatment in CiMist1 mice which lack expression of the Kras G12D mutation in the pancreas, the Treg frequency was increased >twofold in KCiMist1 mice, at a time point when the histopathology of the pancreas shows evidence of pancreatic intraepithelial neoplasia (PanIN) [20]. Thus, this finding suggests a role for malignant cells in directing Treg recruitment to pancreatic tumors (Supplementary Fig. 1).
Using the LSL-Kras G12D/+; LSL-Trp53 R172H/+; Pdx-1Cre (KPC) mouse model of invasive PDAC, we found by flow cytometry a similar result of increased frequency of Foxp3+ Tregs detected in the pancreas without an overt change in the frequency of CD4+ or CD8+ cells among total CD3+ T cells (Fig. 1c and Supplementary Fig. 2). However, since developing PDAC tumors commonly invade or metastasize to peritumoral lymph nodes, we next used microscopy to determine the location of Foxp3+ Tregs that were detected in pancreatic tissue in KPC mice. We found in malignant tissue that the presence of Foxp3+ cells was most pronounced around pancreatic intraepithelial neoplasia (PanIN) which are precursor lesions to the development of invasive PDAC (Fig. 1d, e). However, the majority of Tregs were detected in peritumoral lymph nodes rather than within the tumor bed. The frequency of Tregs detected in peritumoral lymph nodes was similar to non-tumor draining control lymph nodes. Thus, our findings are consistent with Treg recruitment to tumor tissue beginning early during tumorigenesis but with the majority of Tregs remaining confined to lymphatic structures that surround malignant lesions.
Treg depletion and CTLA-4 blockade stimulate CD4 T cell infiltration in PDAC
Tregs are well-recognized for their capacity to dampen T cell immune responses, and in doing so, they can be key proponents of immune escape. Tregs can express high levels of the IL-2 receptor, CD25, which is important for Tregs to maintain an immunosuppressive phenotype [21]. In addition, CTLA-4 is constitutively expressed by Tregs and its expression is critical for Tregs to regulate conventional T cell activation [22] and to modulate suppressive properties of antigen presenting cells [23]. CTLA-4 binds to B7 ligands including CD80 and CD86 molecules that are expressed on antigen presenting cells and has a higher affinity for these ligands than the activating co-receptor CD28 which is essential for activation of conventional naïve T cells [24]. In patients with metastatic breast cancer, CD25 antibodies produce a prolonged decrease in peripheral blood Tregs [21] and in patients with metastatic melanoma, CTLA-4 antibodies induce T cell infiltration into tumors [25]. Therefore, we examined the impact of targeting Tregs, using anti-CD25 and anti-CTLA-4 antibodies, on T cell infiltration into PDAC tumors in KPC mice.
We found that treatment with anti-CD25 versus control reduced the frequency of Foxp3+CD4+ cells in the blood by >75% (Fig. 2a), consistent with prior reports illustrating that this antibody actively reduces Treg frequency in vivo [26]. Although CD25 can also be transiently expressed on conventional T cells upon activation, we found that only a small subset of CD8+ T cells (approximately 5%) expressed CD25 in the peripheral blood (Supplementary Fig. 3a). In addition, the presence of a PDAC tumor did not alter CD25 expression on CD8+ T cells compared to healthy control mice (Supplementary Fig. 3a). Further, anti-CD25 treatment did not produce any significant changes in CD8+ T frequency in the peripheral blood or peritumoral lymph nodes (Supplementary Fig. 3b, c). In contrast to selective Treg depletion seen with anti-CD25 antibodies, treatment with anti-CTLA-4 did not affect the number of Foxp3+CD4+ cells detected in the peripheral blood (Fig. 2b).

Antibodies targeting CD25 and CTLA-4 induce CD4+ T cell infiltration into spontaneous PDAC tumors. Mice (n = 4/group) were treated with: a anti-CD25 (day 1) versus isotype control (day 1) or b anti-CTLA-4 (days 1, 4, 8, 11), or isotype control (days 1, 4, 8, 11). Shown is the impact of treatment with: a anti-CD25 (black) versus control (gray) and b anti-CTLA-4 (black) versus control (gray) on the frequency of CD3+CD4+Foxp3+ Tregs and CD3+CD4+Foxp3neg conventional T cells detected in the peripheral blood after treatment. Shown is: c quantification per high power field (hpf) and d representative images of immunohistochemistry to detect CD3, CD8, CD4 and Foxp3 cells in PDAC tumors from KPC mice at 14 ± 2 days after beginning treatment with anti-CD25 (n = 5) or anti-CTLA-4 (n = 5) with comparison to isotype control (n = 4). Scale at 40× magnification
Within tumor tissue at two weeks after treatment with anti-CD25, we observed a significant increase in CD3+ T cells, representing mainly CD4+ cells, without a change in Foxp3+ cells (Fig. 2c, d). Similarly, we found that administration of an anti-CTLA-4 increased the density of CD4+ T cells detected in PDAC tumor tissue without a significant change in Foxp3+ cells (Fig. 2c, d). In contrast, we found no impact of treatment with anti-CTLA-4 or anti-CD25 antibodies on CD4 T cell presence in pancreata or spleens of healthy control mice (Supplementary Figs. 4 and 5). Unexpectedly, we also found that CTLA-4 and CD25 antibodies produced little impact on CD8+ T cell recruitment to tumors, which is in contrast to reports in metastatic melanoma where significant increases in CD8+ T cells have been observed with CTLA-4 antibodies [25].
We hypothesized that tumor-infiltrating CD4+ T cells might alter the tumor microenvironment through the production of cytokines such as IFN-γ. However, we detected no significant changes in signal transducer and activator of transcription 1 (STAT1) activation which is known to be induced by IFN-γ (Supplementary Fig. 6a, b). Similarly, we observed no changes in the IFN-γ regulated immune checkpoint molecule PD-L1 in the tumor microenvironment (Supplementary Fig. 6a, b) which we have found is rapidly induced on PDAC cells within hours after IFN-γ treatment in vitro, and is more rapidly upregulated on malignant cells than major histocompatibility molecules which are necessary for antigen presentation (Supplementary Fig. 7). In addition, we observed no significant change in the density of the extracellular matrix that surrounds PDAC (Supplementary Fig. 6c, d) which has been previously shown in the KPC model to be depleted with adoptive T cell therapy [27]. Nonetheless, our data show a role for CTLA-4 and CD25 antibodies in regulating CD4+ T cell infiltration into tumors. As treatment did not alter the frequency of Foxp3+ cells in tumors, our findings suggest an inhibitory mechanism mediated by CTLA-4 that occurs outside of the tumor microenvironment.
CD4+ T cell infiltration is regulated by the CTLA-4/CD80 pathway
To understand the mechanism by which either Treg depletion or disruption of CTLA-4 stimulates CD4+ T cell infiltration into PDAC tumors, we next examined antigen presenting cells (APCs) in peritumoral lymph nodes of KPC mice for expression of the CTLA-4 ligands. We found that expression levels of CD80 and CD86 were highest on CD11c+F4/80neg dendritic cells (DC) compared to CD11b+Gr-1+ immature myeloid cells and F4/80+CD11cnegGr-1neg macrophages (Fig. 3a, b). Of the two CTLA-4 ligands, CD80 was most frequently expressed among CD11c+F4/80neg DC (Fig. 3c).

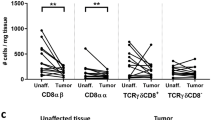
T cell infiltration in PDAC is regulated by CTLA-4/CD80. Shown is expression of: a CD80, and b CD86 on dendritic cells (CD11c+ F4/80neg), granulocytes (CD11b+ Gr-1+), and macrophages (F4/80+ CD11cneg Gr-1neg) from peritumoral lymph nodes in KPC mice (n = 7). c Comparison of CD80 and CD86 expression frequency on CD11c+F4/80neg cells in the lymph node. d Shown are representative immunohistochemistry images of peritumoral lymph nodes from KPC mice at 14 ± 2 days after treatment with control or anti-CD25. Tissues are co-stained for CD11c (brown) and Foxp3 (black). e Quantification of Foxp3+ Treg per high power field (hpf) in peritumoral lymph nodes of control (n = 3) and anti-CD25 (n = 5) antibody-treated KPC mice. f Quantification per hpf and g representative immunohistochemistry images of CD3, CD4, CD8 and Foxp3 cells detected in PDAC tumors from KPC mice (n = 3–5 mice/group) treated with anti-CD80 antibody versus control. Statistical significance was determined by one-way ANOVA and Tukey’s post hoc test. *p < 0.05; **p < 0.01; ***p < 0.001
Using immunohistochemistry, we found that CD11c+ cells co-localized with Foxp3+ cells in peritumoral lymph nodes (Fig. 3d). Moreover, mice treated with the anti-CD25 antibody exhibited an approximate 50% reduction in Foxp3+ Tregs in lymph nodes that was associated with fewer Tregs detected in contact with CD11c+ cells (Fig. 3d, e). Since both CD25 and CTLA-4 antibodies induced CD4 T cell infiltration into PDAC tumors, this finding suggested that CTLA-4 on Tregs may engage with CD80 molecules expressed on DCs in peritumoral lymph nodes to regulate CD4+ T cell infiltration into tumors. Consistent with this hypothesis, anti-CD80 blocking antibodies also induced CD4 T cell infiltration into PDAC tumors (Fig. 3f, g). Together, our findings show a role for the CTLA-4/CD80 pathway regulated by cross-talk between Tregs and DC in peritumoral lymph nodes for controlling CD4 T cell infiltration in PDAC. In addition, our findings suggest that distinct mechanisms regulate CD4+ and CD8+ T cell immunosurveillance in this disease.
Discussion
CTLA-4 antibodies have not produced significant clinical benefit in patients with advanced PDAC. Thus, it is unclear whether CTLA-4 is a relevant therapeutic target for this disease. In this study, we used a clinically relevant mouse model of PDAC to study the role of CTLA-4 antibodies in regulating T cell immunosurveillance. Similar to patients, CTLA-4 antibodies have not demonstrated anti-tumor activity in this model [28]. However, we found that anti-CTLA-4 treatment stimulated CD4+ T cell infiltration into tumors, indicating a role for CTLA-4 in regulating T cell exclusion. This finding was reproducible using CD25 antibodies to deplete Tregs and CD80 antibodies to disrupt CD80 interaction with CTLA-4. However, unlike findings in metastatic melanoma where CTLA-4 antibodies have induced significant increases in tumor-infiltrating CD8+ T cells with evidence of T cell activation [25], we found no significant change in CD8+ T cell recruitment or impact on the tumor microenvironment by infiltrating CD4+ T cells. Our findings suggest that CTLA-4 interacts with CD80 to regulate T cell immunosurveillance but also emphasize that additional mechanisms control CD4+ T cell activation within tumors and the recruitment of CD8+ effector T cells.
The interaction between CTLA-4 on Tregs and CD80/CD86 on APCs can render APCs less potent for stimulating conventional T cell activation [29]. The mechanistic basis for this biology is thought to involve trans-endocytosis in which Tregs use CTLA-4 to capture CD80 and CD86 molecules expressed on APCs [30]. In our studies, we found strong co-localization of Tregs and CD11c+ APCs in peritumoral lymph nodes. However, blockade of CD80 was also capable of inducing T cell infiltration into tumors, which implies a direct immunosuppressive role for CTLA-4/CD80 interactions in regulating T cell infiltration. Consistent with this hypothesis, CD80 expression by DCs has been shown to support Treg-dependent suppression of allogeneic T cell responses [31].
Both CD80 and CD86 are expressed on APCs and can provide co-stimulatory signals to conventional T cells via the activating receptor CD28. These ligands, though, likely do not serve completely redundant functions. For example, CD80 has been shown to be effective in stimulating immunosuppressive activity by Tregs [12]. In contrast, CD86 has been suggested to inhibit Treg suppressive function [31]. Thus, in the absence of CD80 signaling, CD86 may serve to suppress Treg activity while also providing necessary co-stimulation via CD28 to conventional T cells. This would suggest that CD80 and CTLA-4 blocking antibodies disrupt critical Treg interactions with DCs that are necessary for Tregs to suppress conventional CD4+ T cells [32]. It is also possible though that Tregs directly induce APCs with suppressive properties that is dependent on CTLA-4/CD80 signaling [33].
Although CD4+ T cells can exert anti-tumor activity through multiple mechanisms, we previously demonstrated a requirement for both CD4+ and CD8+ T cells in anti-tumor immunity against PDAC [34]. In this prior study, we identified differential regulation of CD4+ and CD8+ T cell infiltration into PDAC that was dependent on phagocytic cells residing outside of tumors. Our results with CTLA-4, CD25, and CD80 antibodies also support a role for immune regulation occurring outside of the tumor microenvironment. For example, we found that CD25 antibodies produced a marked reduction in Treg frequency in peritumoral lymphoid structures but not within PDAC tumors. Thus, manipulating immunoregulatory elements outside of tumors may be critical to enhancing PDAC immunogenicity and improving responsiveness to immunotherapy.
Finally, we undertook this study to investigate the immunological impact of CTLA-4 blockade on the tumor microenvironment in PDAC. Despite lack of clinical activity with CTLA-4 antibodies in PDAC patients, we hypothesized that CTLA-4 antibodies might significantly alter the immune microenvironment as has been seen in metastatic melanoma, where therapy induces T cell activation and infiltration into tumors regardless of treatment response [25]. However, our study shows that CTLA-4 is not a master regulator of T cell exclusion in cancer. This finding may reflect the poor antigenicity of PDAC which contrasts melanoma where high somatic mutational burden is common [35]. In the absence of sufficient antigenic stimulus, CTLA-4 dependent regulation of T cell priming may not be the rate limiting step in the development of tumor-specific T cell immunity. Rather, defects in antigen release and presentation may be more critical for igniting tumor-specific T cell responses. This would be consistent with our prior findings showing that combination of chemotherapy with a CD40 agonist to induce antigen release and APC activation, respectively, is necessary to stimulate productive T cell responses against PDAC in the KPC model [34]. Thus, strategic alignment of multiple immunotherapies aimed at restoring elements of the cancer immunity cycle may be necessary to effectively invoke productive T cell immunity against PDAC [7, 36].
Abbreviations
- CiMist1 :
-
Mist1CreERT2
- CTLA-4:
-
Cytotoxic lymphocyte-associated antigen-4
- DAB:
-
3,3′-Diaminobenzidine
- DC:
-
Dendritic cell
- KCiMist1 :
-
Mist1CreERT2;LSL-Kras G12D/+
- KPC:
-
LSL-Kras G12D/+;LSL-Trp53 R172H/+; Pdx-1Cre
- PanIN:
-
Pancreatic intraepithelial neoplasia
- PC:
-
LSL-Trp53 R172H/+;Pdx-1Cre
- PDAC:
-
Pancreatic ductal adenocarcinoma
- Treg:
-
Regulatory T cell
References
Beatty GL, O’Hara MH, Nelson AM et al (2015) Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. J Clin Oncol 33:3007
Beatty GL, Haas AR, Maus MV et al (2014) Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol Res 2:112–120
Le DT, Lutz E, Uram JN et al (2013) Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother 36:382–389
Le DT, Wang-Gillam A, Picozzi V et al (2015) Safety and survival with GVAX pancreas prime and listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 33:1325–1333
Royal RE, Levy C, Turner K et al (2010) Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 33:828–833
Brahmer JR, Tykodi SS, Chow LQ et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465
Beatty GL, Gladney WL (2015) Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 21:687–692
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264
McCoy KD, Le Gros G (1999) The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol 77:1–10
Schadendorf D, Hodi FS, Robert C et al (2015) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33:1889–1894
Aglietta M, Barone C, Sawyer MB et al (2014) A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol 25:1750–1755
Habbe N, Shi G, Meguid RA et al (2008) Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci USA 105:18913–18918
Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7:469–483
Beatty GL, Chiorean EG, Fishman MP et al (2011) CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331:1612–1616
Uhlen M, Fagerberg L, Hallstrom BM et al (2015) Proteomics. Tissue-based map of the human proteome. Science 347:1260419
Uhlen M, Bjorling E, Agaton C et al (2005) A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteom 4:1920–1932
Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL (2016) IFNgamma and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov 6:400–413
Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, Roncarolo MG, Levings MK (2007) Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol 19:345–354
Devaud C, Darcy PK, Kershaw MH (2014) Foxp3 expression in T regulatory cells and other cell lineages. Cancer Immunol Immunother 63:869–876
McAllister F, Bailey JM, Alsina J et al (2014) Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25:621–637
Rech AJ, Mick R, Martin S et al (2012) CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 4:134
Schmidt EM, Wang CJ, Ryan GA et al (2009) Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J Immunol 182:274–282
Grohmann U, Orabona C, Fallarino F et al (2002) CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol 3:1097–1101
Teft WA, Kirchhof MG, Madrenas J (2006) A molecular perspective of CTLA-4 function. Annu Rev Immunol 24:65–97
Huang RR, Jalil J, Economou JS et al (2011) CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin Cancer Res 17:4101–4109
Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E (1999) Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res 59:3128–3133
Stromnes IM, Schmitt TM, Hulbert A et al (2015) T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 28:638–652
Feig C, Jones JO, Kraman M et al (2013) Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA 110:20212–20217
Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S (2008) Foxp3 + natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA 105:10113–10118
Qureshi OS, Zheng Y, Nakamura K et al (2011) Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332:600–603
Zheng Y, Manzotti CN, Liu M, Burke F, Mead KI, Sansom DM (2004) CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol 172:2778–2784
Matheu MP, Othy S, Greenberg ML et al (2015) Imaging regulatory T cell dynamics and CTLA4-mediated suppression of T cell priming. Nat Commun 6:6219
Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR (2014) Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest 124:2425–2440
Beatty GL, Winograd R, Evans RA et al (2015) Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C (low) F4/80(+) extratumoral macrophages. Gastroenterology 149:201–210
Alexandrov LB, Nik-Zainal S, Wedge DC et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421
Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39:1–10
Acknowledgements
The authors thank Patrick Guirnalda for helpful discussion and Adam Bedenbaugh for advice and technical assistance with immunohistochemistry assays.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Financial support
Support for this project was provided by the following grants from the National Institutes of Health and National Cancer Institute: K08 CA138907 (Gregory L. Beatty) and R01 CA197916 (Gregory L. Beatty). We are grateful to the Molecular Biology and Molecular Pathology and Imaging Cores of the Penn Center supported by a Molecular Studies in Digestive and Liver Diseases grant from the National Institutes of Health. This work was also supported by the following foundations and agencies: AACR-PanCAN Career Development Award (Florencia McAllister), National Pancreas Foundation (Florencia McAllister), Department of Defense Discovery Award (Gregory Beatty), and the Damon Runyon Cancer Research Foundation Innovation Award supported by the Nadia’s Gift Foundation (Gregory Beatty).
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bengsch, F., Knoblock, D.M., Liu, A. et al. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother 66, 1609–1617 (2017). https://doi.org/10.1007/s00262-017-2053-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-017-2053-4