
Abstract
While inflammation has been associated with the development and progression of colorectal cancer, the exact role of the inflammatory Th17 pathway remains unclear. In this study, we aimed to determine the relative importance of IL-17A and the master regulator of the Th17 pathway, the transcription factor RORγt, in the sporadic intestinal neoplasia of APCMIN/+ mice and in human colorectal cancer. We show that levels of IL-17A are increased in human colon cancer as compared to adjacent uninvolved colon. Similarly, naïve helper T cells from colorectal cancer patients are more inducible into the Th17 pathway. Furthermore, IL-17A, IL-21, IL-22, and IL-23 are all demonstrated to be directly mitogenic to human colorectal cancer cell lines. Nevertheless, deficiency of IL-17A but not RORγt is associated with decreased spontaneous intestinal tumorigenesis in the APCMIN/+ mouse model, despite the fact that helper T cells from RORγt-deficient APCMIN/+ mice do not secrete IL-17A when subjected to Th17-polarizing conditions and that Il17a expression is decreased in the intestine of RORγt-deficient APCMIN/+ mice. Differential expression of Th17-associated cytokines between IL-17A-deficient and RORγt-deficient APCMIN/+ mice may explain the difference in adenoma development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is widely accepted that inflammation contributes to the development of cancer [1]. Pathologists have long noted the presence of inflammatory cells in tumors [2]. A relationship between inflammation and intestinal neoplasia is supported by the facts that inflammatory bowel disease predisposes patients to intestinal carcinomas and that the anti-inflammatory drugs aspirin [3–5], celecoxib [6], and rofecoxib [7], all have proven efficacy in preventing human colorectal adenoma development.
The APCMIN/+ mouse strain is the most widely studied murine model of intestinal neoplasia. This model mimics the human condition familial adenoma polyposis (FAP) characterized by the sporadic development of numerous intestinal adenomatous polyps. In the mouse, the small intestine is most affected, whereas the large intestine is most affected in humans. In APCMIN/+ mice, as in humans, a germline mutation in the adenomatous polyposis coli (Apc) gene is responsible for the phenotype.
Despite the fact that adenoma development is controlled by a mutation in a tumor suppressor gene, inflammation is critically important for carcinogenesis in murine models with germline Apc mutations. Adenoma formation is increased after adoptive transfer of pro-inflammatory CD4+CD45RBhi lymphocytes [8] and decreased after the adoptive transfer of anti-inflammatory regulatory T cells (Tregs) [9]. Similarly, adenoma formation is decreased in the absence of the adapter molecule MyD88, which is essential for mediating inflammation from bacterial and viral products [10], as well as the inflammatory cytokine IL-6 [11] and the inflammatory chemokine receptor CCR6 [12].
CD4+ T cells polarized to the Th17 pathway as characterized by the secretion of interleukin 17A (IL-17A) as well as IL-17F, IL-21, and IL-22 are essential for the development of several chronic inflammatory diseases such as multiple sclerosis [13], rheumatoid arthritis [14], psoriasis [15], asthma [16], and inflammatory bowel disease (IBD) [17]. The transcription factor RORγt has been identified as the master regulator of polarization of helper T cells toward the Th17 pathway [18].
Several lines of evidence implicate the Th17 pathway in intestinal carcinogenesis [19–28]. The role of the Th17 in cancer, however, is not clear cut as conflicting evidence supports an anti-tumor role for IL-17A [29, 30]. In this study, we aimed to determine the relative importance of IL-17A and the transcription factor RORγt in sporadic intestinal carcinogenesis.
Methods
Human samples
Tissue samples were acquired immediately after resection from patients undergoing surgery for colorectal cancer at VA Boston Healthcare System (VABHS). Blood was also obtained prior to surgery. Written consent was obtained for collection of all specimens. Oral antibiotic bowel preparation and intravenous antibiotic prophylaxis were routinely used. The protocol was approved by the VABHS Institutional Review Board (IRB) (VABHS IRB protocol #2199).
Animals
APCMIN/+ mice (C57BL/6J-ApcMIN/J) and RORγtGFP/GFP (RORγtKO) mice with the green fluorescent protein (GFP) transcript inserted in place of the first exon of the RORγt gene in both alleles (B6.129P2(Cg)-Rorctm2Litt/J), and wild-type C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). IL-17A−/− (IL-17AKO) mice were generously donated from Dale T. Umetsu (Boston Children’s Hospital, Boston, MA, USA) with the permission of Yoichiro Iwakura (Tokyo, Japan) [31]. APCMIN/+ and RORγtGFP/GFP mice were intercrossed to generate RORγtGFP/GFP–APCMIN/+ (RORγKO–APCMIN/+) and RORγtGFP/+–APCMIN/+ (RORγHET–APCMIN/+) mice. APCMIN/+ mice were also crossed with IL17A−/− mice to generate IL17A−/−–APCMIN/+ (IL17KO–APCMIN/+) mice. Mouse genotypes were confirmed by performing PCR on DNA from tail snips using primers and conditions provided by the Jackson Laboratory and Dr. Iwakura. Experimental mice were not routinely littermates or co-housed. The mice were housed and maintained in the specific pathogen-free (SPF) animal research facility of VABHS. The experimental protocols were approved by VABHS Institutional Animal Care and Use Committee (IACUC) (VABHS IACUC protocols #247, 289, and 291).
Cell lines
The murine colon cancer cell line MC38 was provided by Michael T. Lotze of the University of Pittsburgh [32]. Cell lines were grown in RPMI 1640 (Life Technologies, Grand Island, NY, USA) with 10 % fetal bovine serum (FBS) (Life Technologies) and antibiotic–antimycotic (Life Technologies). The human colorectal adenocarcinoma cell lines HCT 116, HT-29, and SW480 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA).
Proliferation assay
Cells were plated at 10,000 cells per well in RPMI 1640 with 10 % FBS in 96-well plates either in the presence or absence of IL-17A, IL-21, IL-22, or IL-23 (R&D Systems, Minneapolis, MN, USA) at 50 ng/mL for 48 h. Cells were then pulsed with 3H-thymidine (0.5 micro Ci/well) for 6 h and harvested, and radioactivity was counted using the LKB Betaplate scintillation counter.
Quantification of polyps
Following euthanasia, the small intestine was dissected from the pylorus to the ileocecal valve. The intestines were cannulated and flushed with phosphate-buffered saline (PBS) to remove fecal material. The cleaned intestine was divided into three equal length segments: duodenum (first segment), jejunum (second), and ileum (third, most distal). Each segment was cut longitudinally and examined under a dissecting microscope at 4× magnification. Polyps were counted and measured at their greatest diameter [12]. Tumor load was determined by multiplying the number of polyps by their measured diameters [25].
Tumor challenge
MC38 cells were cultured until they reached 80 % confluence. 5 × 105 cells in 100 μL of sterile PBS were injected subcutaneously into the right hindquarter of 6-week-old sex-matched C57BL/6J (WT) and RORγKO mice. Tumors were measured by their greatest dimension 3 times a week until they reached an endpoint of 2 cm in largest diameter or ulceration.
Human IL-17A concentration in colon cancer specimens
Snap-frozen sections of human tissue were stored at −80 °C until use. Upon thawing, tissues were homogenized. Homogenates were made in a volume of 200 μL of PBS using an Eppendorf micropestle in a 1 mL Eppendorf tube (Eppendorf, Hauppauge, NY, USA). The homogenized samples were then centrifuged at 13,000 rpm for 10 min to remove debris, and the supernatant was collected. Protein concentration was measured using the Quick Start Bradford protein assay (Bio-Rad Life Sciences, Hercules, CA, USA). IL-17A levels were assessed by multiplexed ELISA using a Q-plex assay chemiluminescent kit (Quansys Bioscience, UT). All buffers/solutions were provided as part of the Quansys 16-plex kit. Briefly, supernatants were diluted as per protocol in sample diluent buffer. Recombinant cytokine was serially diluted to create a standard curve. Fifty microliters of each dilution and 50 μL of diluted test sample supernatants were added to a 96-well Q-Plex Array plate. The plate was incubated for 1 h at room temperature on an orbital shaker (500 rpm). Following incubation, plates were washed 3 times with wash buffer and incubated with 50 mL of detection buffer for 1 h on a shaker (500 rpm). The wells were again washed 3 times with buffer, and 50 μL of 1× Streptavidin–HRP solution was added to each well for 15 min at room temperature on a shaker (500 rpm). Next, the wells were washed 3 times with wash buffer and incubated for 15 min with 50 μL per well of mixed substrates A and B. The plate was then imaged using a Q-View imager (Quansys Bioscience). Results were analyzed using Q-view software (Quansys Bioscience).
Expression data from The Cancer Genome Atlas
Data from The Cancer Genome Atlas (TCGA) for colon adenocarcinoma (COAD) [33] were downloaded from the Broad Institute’s Firehose pipeline using RTCGAToolbox [34]. Agilent microarray-based data were used to compare expression of IL17A in colon cancers and normal colon. The empirical Bayes method for assessing differential expression was used for statistical analysis. Microarray- and RNAseq-based gene-level expression data were used to test the correlation between IL17A and RORC expression using Pearson correlation coefficients. R and the Bioconductor limma package were used for statistical analysis.
RNA extraction and qRT-PCR
Murine small intestinal tissue was dissected from 15-week-old mice and cleaned as was done for quantification of polyps above. Once clean, the third portion of the intestine (ileum) was snap frozen and stored at −80 °C until use. Upon thawing, cell lysates were prepared from ~100 mg of intestinal tissue by addition of 900 µl of QIAzol Lysis Reagent (Qiagen) in an Eppendorf tube followed by tissue homogenization using the Tissue Lyser II (Qiagen). RNA extraction was performed using the RNeasy Plus Universal Kit (Qiagen, Valencia, CA, USA). For cDNA synthesis, 5 µg of total RNA was reverse-transcribed using the SuperScript III First-Strand Synthesis System (Life Technologies) where oligo-dT was used as the first-strand primer. qPCR for Il23, Il22, Il21, Il17a, Il6, Ifng, Tnfa, Il10, Ki67, Bad, Bax, Bbc3 (PUMA), Bcl2, and Bcl2l1 (Bcl-xL) was performed from cDNA. The primers are listed in Supplementary Table 1. Fold change was calculated using the 2-ΔΔCt method with normalization to GAPDH.
Mouse Th17 differentiation
Spleens were harvested from 6- to 8-week-old mice, mechanically dissociated using a 100 µM filter, and washed with RPMI. DNase I (Roche Life Science, Indianapolis, IN, USA) was added (1:200 dilution 50 µg/mL). Red blood cells were lysed using BD Pharm Lyse (BD Biosciences, San Jose, CA, USA). Naïve CD4+ cells were extracted using the CD4+CD62L+ T Cell Isolation Kit II (Miltenyi Biotec, Cologne, Germany). Two rounds of negative selection were performed followed by 1 round of positive selection per the manufacturer’s protocol. A 12-well polystyrene plate was coated with anti-CD3 (clone 145-2C11, eBioscience, San Diego, CA, USA) (4 µg/mL) and anti-CD28 (2 µg/mL) (clone 37.51.1, Invitrogen, Grand Island, NY, USA) diluted in PBS and incubated for 3 h at 37 °C. PBS was removed, and 1 × 106 cells were added to each well along with anti-IL-4 antibody (1:250, clone 11B11, eBioscience), IL-6 (50 ng/mL) (R&D Systems), TGF-β1 (5 ng/mL) (R&D Systems), IL-23 (10 ng/mL) (R&D Systems), anti-IFN-γ antibody (5 μg/mL, clone XMG1.2, eBioscience), and IL-1β (20 ng/mL) (R&D Systems). After 6 days, cells were stimulated with phorbol 12-myristate13-acetate (PMA) (50 ng/mL) and ionomycin (750 ng/mL) for a total of 5 h. Brefeldin A (25 μg/mL) was added (Sigma-Aldrich, St. Louis, MO, USA) after the first hour. Following stimulation, rat anti-mouse CD16/32 (clone 2.4G2, BD Biosciences) was added at a 1:100 dilution for 10 min. Anti-mouse CD3 APC (clone 145-2C11, BD Biosciences) and anti-mouse CD4 AlexaFluor700 (clone GK1.5, eBioscience) were added, and cells were incubated for 20 min. Cells were fixed with 2 % paraformaldehyde for 20 min and then permeabilized with 0.5 % saponin (Sigma-Aldrich) and stained with anti-mouse IL-17A-PE (clone TC11-18H10, BD Biosciences) and anti-mouse IFNγ-PE-Cy7 (clone XMG1.2, BD Biosciences).
Human Th17 differentiation
PBMCs were isolated from whole blood using a Ficoll-Paque gradient (GE Healthcare, Piscataway, NJ, USA). PBMCs were polarized with a cocktail consisting of IL-1β (10 ng/mL), IL-6 (20 ng/mL), and IL-23 (10 ng/mL) in addition to anti-IFN-γ (10 µg/mL), anti-IL-4 (10 µg/mL), TGF-β (1 ng/mL) (all from R&D Systems) and anti-CD3 and anti-CD28 antibodies (BD Biosciences) for 6 days in RPMI 1640 supplemented with 10 % FBS. Cells were further expanded with IL-2 (20 U/mL; R&D Systems) for an additional 6 days prior to restimulation with PMA and ionomycin (Sigma) for 6 h in the presence of GolgiStop (eBioscience). The cells were stained for CD4 (BD Biosciences), and after washing, cells were fixed with Cytofix/Cytoperm buffer (BD Biosciences). Intracellular staining for IFN-γ and IL-17A was performed using conjugated anti-IFN-γ (BD Biosciences) and anti-IL-17A (eBioscience) antibodies and analyzed using flow cytometer (BD Biosciences) as described previously [35].
Results
As several reports suggest that the Th17 pathway may promote intestinal carcinogenesis, we first sought to assess the expression of IL-17A in human colorectal cancer. IL-17A protein levels were measured in matched samples of human colorectal cancer tumors and adjacent uninvolved colon. The mean level of IL-17A expression was found to be significantly greater in the tumors. Of the 19 matched samples analyzed, there was more IL-17A detected in the tumor in 13 (68 %) with less noted in only 3 (16 %). In 10 matched samples (53 %), the tumor had at least double the level of IL-17A. The magnitude of increase in IL-17A level ranged up to over 47,000 % (Fig. 1a). An increase in IL17A gene expression was confirmed using data from The Cancer Genome Atlas (TCGA) (Fig. 1b). A more modest increase in IL17A expression in colon cancers as compared to non-diseased colon was seen (fold change 1.14, p = 0.01). In contrast, expression of RORC, the gene encoding RORγt, the prototypical transcription factor that drives Th17 differentiation, was decreased in tumors as compared to non-diseased colon (fold change 1.7, p < 0.001). (Supplementary Fig. 1). Interestingly, a correlation between IL17A and RORC expression was not found (Supplementary Fig. 2).

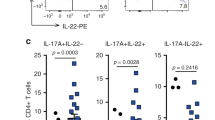
IL-17A levels are overexpressed in human colon cancers, and helper T cells from human colorectal cancer patients are more inducible into the Th17 pathway. a IL-17A expression in samples of human colon cancer and matched adjacent normal tissue (n = 19) was assayed by ELISA and normalized to total protein measured by the Bradford assay. The values for cancer and normal tissue were compared for each patient, and the Wilcoxon signed-rank test was used to assess significance. b IL17A gene expression in 153 colon cancers and 19 normal colon samples was determined from microarray data. The boxes represent the interquartile range (IQR); the lines represents the median; the error bars represent the lowest and highest data still within 1.5 IQR of the upper and lower quartiles, respectively; and the circles represent the data points outside of 1.5 IQR of the upper and lower quartiles. The empirical Bayes method for assessing differential expression was used for statistical analysis. c PBMCs were isolated from normal donors and colon cancer patients and then activated with a Th17-polarizing cocktail for 12 days. Intracellular IL-17A and IFN-γ were measured by flow cytometry following PMA/ionomycin restimulation. The right panels are representative dot plot analyses from one healthy patient and one patient with stage IV colon cancer showing the percentage of cells that are positive for intracellular IL-17A or IFN-γ within the gated CD4+ population. The cells were single-stained for each cytokine. Composite results are presented in the bar graph on the left as mean values with standard error of the mean (*p < 0.05, compared to healthy controls). The Student’s t test was used for statistical comparison
We next assessed whether helper T cells in colorectal cancer patients were more prone to be induced into the Th17 pathway. PBMCs from colorectal cancer patients and healthy controls were purified and subjected to Th17-polarizing conditions and then non-specifically stimulated and analyzed by flow cytometry with intracellular staining. The mean number of CD4+ cells expressing IL-17A after polarization was approximately 7.5-fold higher in the cancer patients as compared to healthy controls. In contrast, the level of IFN-γ-expressing CD4+ cells was approximately 2.5-fold lower in the cancer patients. We did not note a significant variation with stage in the percent of CD4+ cells expressing either IL-17A or IFN-γ (Fig. 1c).
We then sought to explore a causal relationship between the Th17 pathway and tumor growth. We first examined whether cytokines associated with the Th17 pathway were directly mitogenic to colorectal cancer cells. We subjected 3 different human colorectal cancer cell lines to exposure to IL-17A, IL-21, IL-22, and IL-23 and measured proliferation. Two of the cell lines (Hct 116 and HT-29) appeared responsive to the Th17 cytokines as exposure to each of the 4 cytokines significantly increased proliferation from 203 to 879 %. Proliferation of the third cell line (SW480) was not affected by IL-17A, IL-21, IL-22, or IL-23 (Fig. 2).

Human colorectal cancer cell lines proliferate in response to Th17 cytokines. Proliferation of cell lines cultured with or without 50 ng/mL of IL-17A, IL-21, IL-22, or IL-23 for 48 h was assessed by thymidine incorporation. Thymidine incorporation of cells cultured with cytokines is shown as a percentage of that for the same cell line cultured without cytokine. Error bars show the standard error of the mean (*p < 0.05, compared to thymidine incorporation in the absence of cytokine). Data are representative of 3 independent experiments. The Student’s t test was used for statistical comparisons
To further assess the role of the Th17 pathway in intestinal tumorigenesis, we used a well-validated model of spontaneous intestinal tumorigenesis that is driven by loss of a tumor suppressor gene. APCMIN/+ or multiple intestinal neoplasia (MIN) mice are heterozygous for a germline mutation in the adenomatous polyposis coli (Apc) gene, resulting in a non-functional gene product. Mice spontaneously develop multiple intestinal adenomas mimicking the human condition FAP. We bred these animals to mice deficient in RORγt (RORγtKO mice), to create APCMIN/+ mice that were either heterozygous or homozygous for loss of RORγt (RORγtHET–APCMIN/+ mice and RORγtKO–APCMIN/+ mice, respectively). We first confirmed that helper T cells from RORγtKO–APCMIN/+ do not secrete IL-17A when subjected to Th17-polarizing conditions. While the number of IFN-γ-expressing cells after activation with a Th17-polarizing cocktail was similar between the different genotypes, there were minimal IL-17A-expressing cells from the IL-17A knockout (IL-17AKO), RORγtKO, and RORγtKO–APCMIN/+ mice while IL-17A production was maintained in the WT and APCMIN/+ mice (Fig. 3).

Helper T cells from RORγtKO–APCMIN/+ do not produce IL-17A when subjected to Th17-polarizing conditions. Naïve CD4+ T cells were purified from the spleens of 6- to 8-week-old mice from WT, IL-17AKO, RORγtKO, APCMIN/+, and RORγtKO–APCMIN/+ mice using magnetic column purification. Cells were cultured in anti-CD3/anti-CD28 coated wells along with anti-IL-4, IL-6, TGF-β, IL-23, anti-IFN-γ, and IL-1β for 6 days and then stimulated with PMA and ionomycin followed by Golgi blockade with brefeldin A. Cells were then analyzed by flow cytometry after intracellular staining for IL-17A and IFN-γ. a Representative flow cytometry data. b The percent of cells expressing IL-17A were compared. p = 0.05 by the Student’s t test. Data are derived from 5 independent experiments (n = 5 of each APCMIN/+ and RORγtKO–APCMIN/+ mice)
We then compared polyp development in APCMIN/+, IL-17AKO–APCMIN/+, RORγtHET–APCMIN/+, and RORγtKO–APCMIN/+ mice. At 15 weeks of age, the total polyp number and total polyp load in the small intestine were significantly decreased for IL-17AKO–APCMIN/+ mice as compared to APCMIN/+ mice (Fig. 4a). The polyp number and load were also found to be significantly decreased in each segment of the small bowel (duodenum, jejunum, and ileum; Fig. 4b). In contrast, polyp number and load were preserved for both the RORγtHET–APCMIN/+ and RORγtKO–APCMIN/+ mice in the entire small intestine and in each segment (Fig. 4). Polyps were also quantified for RORγtHET–APCMIN/+ and RORγtKO–APCMIN/+ mice at 22 weeks, and no difference was found in the polyp number overall or in individual segments. A very small but significant decrease in polyp load was seen in RORγtKO–APCMIN/+ mice that were restricted to the ileum (Supplementary Fig. 3). We assessed relative expression of genes associated with proliferation and apoptosis in the adenoma-bearing intestines of APCMIN/+, RORγtKO–APCMIN/+, and IL-17AKO–APCMIN/+ mice. The proliferation gene Ki67 was modestly downregulated in the intestines of RORγtKO–APCMIN as compared to APCMIN mice, whereas expression did not appear to be altered in the intestines of IL-17AKO–APCMIN/+ mice (Supplementary Fig. 4a). In contrast, adenoma-bearing intestine from IL-17AKO–APCMIN/+ mice had decreased expression of the anti-apoptotic genes Bad, Bax, and Bbc3 (PUMA) with increased expression of the pro-apoptotic genes Bcl2 and Bcl2l1 (Bcl-xL) in comparison with APCMIN mice, whereas the expression of these genes was only altered modestly if at all in RORγtKO–APCMIN/+ mice (Supplementary Fig. 4b).

Lack of IL-17A but not RORγt is associated with decreased development of intestinal adenomas. Polyp number and polyp load of 15-week-old mice are shown for a the entire small intestine and b the duodenum, jejunum and ileum individually (n = 7–12 mice per group) (*p < 0.05 compared to APCMIN/+ mice, † p < 0.05 compared to RORγtHET–APCMIN/+ mice, ‡ p < 0.05 compared to RORγtKO–APCMIN/+ mice). Error bars show the standard error of the mean. The Student’s t test was used for statistical comparisons
We also explored whether deficiency of RORγt affected tumor growth after subcutaneous injection of the colorectal cancer cell line MC38, which is syngeneic for C57/Bl6 mice. In this model, we did not detect any difference in tumor growth or in survival between WT and RORγtKO mice after tumor challenge (Fig. 5).

Lack of RORγt does not affect tumor growth or survival after subcutaneous implantation of a syngeneic colon cancer. a Mean maximal tumor diameter and b survival versus time are shown after injection of 5 × 105 MC38 cells subcutaneously into the flank of WT and RORγtKO mice (n = 5–6 per group). Mice were euthanized if they reached endpoints of a maximal tumor diameter of 2 cm or ulceration. Data are representative of 2 independent experiments. One-way analysis of covariance for independent samples (ANCOVA) and the log-rank test were used to assess significance of tumor growth and survival, respectively
Lastly, in order to attempt to explain the difference in intestinal tumorigenesis observed between RORγtKO–APCMIN/+ and IL-17AKO–APCMIN/+ mice, we assessed the relative expression of cytokines in the adenoma-bearing intestines of APCMIN/+, RORγtKO–APCMIN/+, and IL-17AKO–APCMIN/+ mice. The inflammatory cytokines Il23, Il17a, and Il6 were decreased in the intestines of RORγtKO–APCMIN/+ and IL-17AKO–APCMIN/+ mice as compared to those of APCMIN/+ mice. The magnitude of the decrease in Il17a expression was greater in the IL-17AKO–APCMIN/+ mice. There appeared to be alterations in the expression of Il22 and Il21 between the intestines of RORγtKO–APCMIN/+ and IL-17AKO–APCMIN/+ mice as compared to those of APCMIN/+ mice. While these cytokines were both decreased in RORγtKO–APCMIN/+ mice intestines, Il22 was only marginally decreased and Il21 was increased in the IL-17AKO–APCMIN/+ intestines (Fig. 6). As IL-21 can be associated with increased tumor immunity through granzyme B- and perforin-dependent mechanisms [36], we also assessed expression of Gzmb1 and Prf1. Consistent with the hypothesis that the increased Il21 expression in the intestine of IL-17AKO–APCMIN/+ mice may contribute to the decreased development of intestinal adenomas, we also noted that the expression of Gzmb1 and Prf1 appeared to mirror the expression of Il21 in the intestines of RORγtKO–APCMIN/+ and IL-17AKO–APCMIN/+ mice (Supplementary Fig. 5). We did not see marked differences in the expression of Ifng, Tnfa, and Il10 in the intestine of RORγtKO–APCMIN/+ versus IL-17AKO–APCMIN/+ mice (Fig. 6).

Relative cytokine expression in adenoma-bearing intestine from APCMIN/+, RORγtKO–APCMIN/+, and IL-17AKO–APCMIN/+ mice. Ileum of 15-week-old mice was lysed and RNA extracted. cDNA was then synthesized. qRT-PCR was performed. The relative quantification of gene expression was calculated. The fold difference in gene expression for RORγtKO–APCMIN/+ and IL-17AKO–APCMIN/+ mice relative to APCMIN/+ mice is shown on a log2 scale. Bars above the horizontal axis represent increased expression relative to APCMIN/+ mice, while those below the horizontal axis represent decreased expression relative to APCMIN/+ mice
Discussion
The aim of this paper was to determine the relative importance of IL-17A and RORγt in sporadic intestinal carcinogenesis. We have demonstrated that IL-17A is overexpressed in human colorectal cancer and that peripheral helper T cells from colorectal cancer patients are more inducible into the Th17 pathway. We have also shown that IL-17A, IL-21, IL-22, and IL-23, cytokines associated with the Th17 pathway, are directly mitogenic to human colorectal cancer cell lines. Interestingly, however, deficiency of IL-17A but not RORγt, the master regulator of the Th17 pathway, is associated with decreased murine sporadic intestinal carcinogenesis. Similarly, deficiency of RORγt does not impair tumor growth in a syngeneic mouse adoptive transplant model. Lastly, we explored the cytokine milieu of the adenoma-bearing intestines to better understand the different phenotype between the IL-17A- and RORγt-deficient APCMIN/+ mouse models. The inflammatory cytokines Il17a and Il23 as well as Tnfa and Il6 were all suppressed in both models. Unexpectedly, though, expression of Il21 was increased in the intestine of IL-17A-deficient APCMIN/+ mice, while it was decreased in RORγt-deficient APCMIN/+ mice.
Our finding of the immune system being skewed toward the Th17 pathway in colorectal cancer is supported by other studies. For example, our finding that IL-17A is upregulated at the protein level in human colorectal carcinomas is consistent with the finding that IL17A [19, 20, 25] and IL23 [25] are upregulated at the mRNA level. While an increased infiltration of T cells expressing IL-17A infiltrating colorectal tumors as detected by immunohistochemistry has been previously reported [20], to our knowledge it has not been previously demonstrated that peripheral blood helper T cells from colorectal cancer patients are more apt to be induced to the Th17 pathway. Interestingly, however, we noted that RORC, the gene encoding RORγt, did not appear to be upregulated in human colon cancer and that tumor expression of RORC did not correlate with that of IL17A, possibly suggesting that RORγt may not be as significant in colon cancer and may not account for the increased IL-17A.
While a link of inflammation with the development and progression of colorectal cancer has been established, the mechanisms behind this relationship are poorly understood. Attention has been focused on the importance of the Th17 pathway. In mouse models of sporadic intestinal carcinogenesis, the Th17-associated cytokines IL-17A [22, 25], IL-17F [23], IL-22 [27], and IL-23 [25, 28] have all been shown to promote tumor development even in the absence of other frank inflammatory pathologies. It has been suggested that effect of IL-23 in promoting intestinal neoplasia may be at least partially mediated through IL-17A [25, 28]. We are not aware of other data demonstrating a direct mitogenic effect of the Th17-associated cytokines IL-17A, IL-21, IL-22, and IL-23 on neoplastic epithelial cells in colorectal cancers. This has been shown, however, for related cytokines such as IL-6 and TNF-α [37].
In this study, we confirm that deficiency of IL-17A in APCMIN/+ mice results in decreased spontaneous intestinal tumorigenesis [22]. Surprisingly, however, we found that deficiency of RORγt, the master regulator of the Th17 pathway, does not reduce adenoma development in the same model. We also did not find reduced growth of transplanted MC38 syngeneic mouse colon cancer tumors. Of note, growth of MC38 in the same adoptive transplant model was not impacted by host deficiency of IL-17A in one study [38], despite the fact that another study using this model showed that abrogation of IL-17A with an adenovirus siRNA vector decreased growth of MC38 [39]. Perhaps deficiency of host IL-17A without ablating any contribution of IL-17A from tumor cells is not sufficient to reduce tumor growth.
Our finding of a lack of effect of RORγt deficiency on sporadic intestinal polyposis appears to conflict with another study that reported decreased adenomas in the setting of RORγt deficiency in a similar mouse model of sporadic intestinal tumorigenesis induced by an Apc mutation [26]. The model used in the conflicting study, APCΔ468 mice, has 1 allele of the Apc gene with exons 11 and 12 deleted, resulting in a prematurely terminated 468 amino acid protein [40]. In contrast, APCMIN/+ mice have 1 Apc allele with a T to A transversion at nucleotide 2549, resulting in a premature stop codon at codon 850 [41]. While it is not clear how the shorter Apc truncation in the other study could result in such a different result, there is a previous instance of a dramatic difference between these 2 models relating to the influence of the immune system on tumorigenesis. A study using APCΔ468 mice reported that mast cells promote adenoma development [42], whereas a separate study using APCMIN/+ mice demonstrated that mast cells exert a protective effect against polyposis [43] despite the fact that both studies used mast cell-deficient KitW−sh/Wsh (SASH) mice on backgrounds of the respective Apc mutant animals. Further study will be needed to determine which of these mouse systems better models human colorectal carcinogenesis.
It is not obvious why APCMIN/+ mice deficient in IL-17A have dramatically decreased intestinal tumorigenesis while those deficient in RORγt do not. Despite the fact that RORγt is the most important transcriptional regulator of IL-17A expression in helper T cells [44] and that helper T cells from RORγt-deficient mice do not polarize into Th17 cells [18], it is conceivable that deficiency of RORγt might not abrogate Th17 polarization in APCMIN/+ mice. Nevertheless, we have shown that helper T cells from RORγt-deficient APCMIN/+ mice, as expected, do not secrete IL-17A after exposure to Th17-polarizing conditions. It is also possible that despite the absence of RORγt, IL-17A might still be expressed in the adenoma-bearing intestine of APCMIN/+ mice as IL-17A can be secreted by cells other than the helper T cells that were assayed in the polarization assays and as RORγt-independent secretion of IL-17A has been described [45]. While Il17a expression is dramatically reduced in the adenoma-bearing intestine of RORγt-deficient APCMIN/+ mice, it may not be decreased to the same extent as IL-17A-deficient APCMIN/+ mice. It is possible that a threshold reduction of IL-17A is required for a reduction in adenoma growth. The concept of RORγt-independent secretion of IL-17A in colorectal cancer may be supported by the fact that RORC is actually decreased in human colon tumors despite the increased expression of IL-17A and that expression of RORC and IL17A in tumors is not correlated.
Another theoretic possibility is that an effect of RORγt deficiency other than loss of IL-17A expression is responsible for the different phenotypes. RORγt is involved in several immune functions beyond the development of classical Th17 cells. For example, RORγt-deficient mice fail to develop lymphoid organs such as lymph nodes and Peyer’s patches [46]. RORγt is also critical for the development and function of innate lymphoid cells (ILCs) [47, 48]. In addition, RORγt is involved in the development of other immune cell subsets including subsets of γδ T cells [49] and NKT cells [50]. There is considerable plasticity between Tregs and Th17 cells, and in fact, RORγt may be involved in the development of Tregs, including Tregs that subsequently turn off RORγt and do not generate IL-17A [51]. Despite the multifaceted role of RORγt in the immune system, loss of RORγt eliminates other IL-17A-mediated pathology in the gut [52].
An intriguing possibility is that another alteration in cytokine production may counterbalance the loss of IL-17A. In the panel of cytokines that we assessed, we did not find differences in expression of the prototypical Th1 cytokine Ifng or the regulatory cytokine Il10 in the adenoma-bearing intestine of IL-17A-deficient or RORγt-deficient APCMIN/+ mice. We also did not see differences in the expression of the inflammatory cytokines Il23, Tnfa, or Il6. We did, however, detect upregulation of Il21 in the IL-17A-deficient animals as compared to downregulation of Il21 in the RORγt-deficient animals. As IL-21 can elicit anti-tumor immune responses [36], it is conceivable that the decreased Il21 in RORγt-deficient APCMIN/+ mice may contribute to the difference in intestinal tumorigenesis. The differences in the levels of Prf1 (perforin) and Gzmb1 (granzyme B) in the 2 models also support this hypothesis. Further experimentation is needed, however, to definitively determine whether the alterations in Il21 expression explain the difference in adenoma growth and to delineate the mechanism of this effect.
Interestingly, the cytokine expression in the adenoma environment of the different mouse models corresponds to the expression of the proliferation gene Ki67 as would be expected based on the effect of the cytokines on proliferation seen in vitro. APCMIN/+ mice deficient in RORγt have modestly decreased intestinal expression of Ki67 consistent with the decreased expression of the cytokines Il17a, Il21, Il22, and Il23, all of which were shown to promote proliferation of colorectal cancer cell lines. In contrast, IL-17A-deficient APCMIN/+ mice, which have decreased intestinal expression of Il17a and Il23 without a change in Il22 expression and with increased Il21, did not appear to have a change in Ki67 expression compared to APCMIN/+ mice. It seems that changes in apoptosis may be more important than changes in proliferation in regulating adenoma growth in these mouse models. Decreased expression of the anti-apoptotic genes Bad, Bax, and Bbc3 (PUMA) and increased expression of the pro-apoptotic genes Bcl2 and Bcl2l1 (Bcl-xL) mirrored the decreased polyp number for IL-17A-deficient APCMIN/+ mice, whereas the expression of these genes was only altered modestly if at all in RORγtKO–APCMIN/+ mice, consistent to what was observed for adenoma growth. These alterations in apoptotic gene expression are also similar to alterations in Gzmb1 and Prf1, which code for proteins secreted by lymphocytes and natural killer cells to induce the apoptosis-mediated cell death of target cells.
In summary, our results suggest that loss of IL-17A but not RORγt diminishes adenoma development in a mouse model of sporadic intestinal carcinogenesis. The lack of measurable change in adenoma development despite the loss of RORγt occurs despite the fact that production of IL-17A is impaired in this model. These data imply that strategies directed against the Th17 pathway in colorectal cancer should focus on inhibition of IL-17A itself rather than on RORγt.
Abbreviations
- ANCOVA:
-
Analysis of covariance for independent samples
- APC:
-
Adenomatous polyposis coli
- Apc :
-
Adenomatous polyposis coli gene
- ATCC:
-
American Type Culture Collection
- FAP:
-
Familial adenomatous polyposis
- HET:
-
Heterozygote
- IACUC:
-
Institutional Animal Care and Use Committee
- IBD:
-
Inflammatory bowel disease
- ILC:
-
Innate lymphoid cell
- ILC3:
-
Group 3 innate lymphoid cell
- IRB:
-
Institutional review board
- KO:
-
Knockout
- MIN:
-
Multiple intestinal neoplasia
- NKT cell:
-
Natural killer T cell
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- siRNA:
-
Small interfering ribonucleic acid
- SPF:
-
Specific pathogen-free
- Treg:
-
Regulatory T cell
- VABHS:
-
VA Boston Healthcare System
- WT:
-
Wild type
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Virchow RC (1863) The malignant neoplasias. In: Hirschwald A (ed) Thirty lectures hold within the winter-semester 1862–1863. Berlin University, Berlin
Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, McKeown-Eyssen G, Summers RW, Rothstein R, Burke CA, Snover DC, Church TR, Allen JI, Beach M, Beck GJ, Bond JH, Byers T, Greenberg ER, Mandel JS, Marcon N, Mott LA, Pearson L, Saibil F, van Stolk RU (2003) A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med 348(10):891–899. doi:10.1056/NEJMoa021735
Benamouzig R, Deyra J, Martin A, Girard B, Jullian E, Piednoir B, Couturier D, Coste T, Little J, Chaussade S (2003) Daily soluble aspirin and prevention of colorectal adenoma recurrence: one-year results of the APACC trial. Gastroenterology 125(2):328–336. doi:10.1016/S0016-5085(03)00887-4
Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi CL, Steinbach G, Schilsky R (2003) A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med 348(10):883–890. doi:10.1056/NEJMoa021633
Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, Boisserie F, Anderson WF, Viner JL, Bagheri D, Burn J, Chung DC, Dewar T, Foley TR, Hoffman N, Macrae F, Pruitt RE, Saltzman JR, Salzberg B, Sylwestrowicz T, Gordon GB, Hawk ET (2006) Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 355(9):873–884. doi:10.1056/NEJMoa061355
Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A, Bolognese JA, Oxenius B, Horgan K, Loftus S, Morton DG (2006) A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology 131(6):1674–1682. doi:10.1053/j.gastro.2006.08.079
Rao VP, Poutahidis T, Ge Z, Nambiar PR, Horwitz BH, Fox JG, Erdman SE (2006) Proinflammatory CD4+CD45RB(hi) lymphocytes promote mammary and intestinal carcinogenesis in Apc(Min/+) mice. Cancer Res 66(1):57–61. doi:10.1158/0008-5472.CAN-05-3445
Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, Schauer DB (2005) CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+mice. Cancer Res 65(10):3998–4004. doi:10.1158/0008-5472.CAN-04-3104
Rakoff-Nahoum S, Medzhitov R (2007) Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 317(5834):124–127. doi:10.1126/science.1140488
Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA (2008) Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol 294(2):R393–R401. doi:10.1152/ajpregu.00716.2007
Nandi B, Pai C, Huang Q, Prabhala RH, Munshi NC, Gold JS (2014) CCR6, the sole receptor for the chemokine CCL20, promotes spontaneous intestinal tumorigenesis. PLoS ONE 9(5):e97566. doi:10.1371/journal.pone.0097566
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421(6924):744–748. doi:10.1038/nature01355
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ (2003) Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 198(12):1951–1957. doi:10.1084/jem.20030896
Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W (2007) Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445(7128):648–651. doi:10.1038/nature05505
Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, Schnyder B (2006) Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med 203(12):2715–2725. doi:10.1084/jem.20061401
Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D (2006) IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 116(5):1310–1316. doi:10.1172/JCI21404
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126(6):1121–1133. doi:10.1016/j.cell.2006.07.035
Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M (2006) IL-23 promotes tumour incidence and growth. Nature 442(7101):461–465. doi:10.1038/nature04808
Le Gouvello S, Bastuji-Garin S, Aloulou N, Mansour H, Chaumette MT, Berrehar F, Seikour A, Charachon A, Karoui M, Leroy K, Farcet JP, Sobhani I (2008) High prevalence of Foxp3 and IL17 in MMR-proficient colorectal carcinomas. Gut 57(6):772–779. doi:10.1136/gut.2007.123794
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15(9):1016–1022. doi:10.1038/nm.2015
Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL (2010) Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci USA 107(12):5540–5544. doi:10.1073/pnas.0912675107
Chae WJ, Bothwell AL (2011) IL-17F deficiency inhibits small intestinal tumorigenesis in ApcMin/+ mice. Biochem Biophys Res Commun 414(1):31–36. doi:10.1016/j.bbrc.2011.09.016
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pages F, Galon J (2011) Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 71(4):1263–1271. doi:10.1158/0008-5472.CAN-10-2907
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M (2012) Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491(7423):254–258. doi:10.1038/nature11465
Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, Ham S, Sandall BP, Khan MW, Mahvi DM, Halverson AL, Stryker SJ, Boller AM, Singal A, Sneed RK, Sarraj B, Ansari MJ, Oft M, Iwakura Y, Zhou L, Bonertz A, Beckhove P, Gounari F, Khazaie K (2012) Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci Transl Med 4(164):164ra159. doi:10.1126/scitranslmed.3004566
Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O’Connor W Jr, Murphy AJ, Valenzuela DM, Yancopoulos GD, Booth CJ, Cho JH, Ouyang W, Abraham C, Flavell RA (2012) IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491(7423):259–263. doi:10.1038/nature11535
Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, Vives F, Moon C, Penaflor E, Turner S, Ayanoglu G, Chang C, Basham B, Mumm JB, Pierce RH, Yearley JH, McClanahan TK, Phillips JH, Cua DJ, Bowman EP, Kastelein RA, LaFace D (2014) Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol 7(4):842–856. doi:10.1038/mi.2013.101
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114(6):1141–1149. doi:10.1182/blood-2009-03-208249
Hinrichs CS, Kaiser A, Paulos CM, Cassard L, Sanchez-Perez L, Heemskerk B, Wrzesinski C, Borman ZA, Muranski P, Restifo NP (2009) Type 17 CD8+ T cells display enhanced antitumor immunity. Blood 114(3):596–599. doi:10.1182/blood-2009-02-203935
Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y (2002) Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17(3):375–387. doi:10.1016/S1074-7613(02)00391-6
Rosenberg SA, Spiess P, Lafreniere R (1986) A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233(4770):1318–1321. doi:10.1126/science.3489291
The Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487(7407):330–337. doi:10.1038/nature11252
Samur MK (2014) RTCGAToolbox: a new tool for exporting TCGA Firehose data. PLoS ONE 9(9):e106397. doi:10.1371/journal.pone.0106397
Prabhala RH, Pelluru D, Fulciniti M, Prabhala HK, Nanjappa P, Song W, Pai C, Amin S, Tai YT, Richardson PG, Ghobrial IM, Treon SP, Daley JF, Anderson KC, Kutok JL, Munshi NC (2010) Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 115(26):5385–5392. doi:10.1182/blood-2009-10-246660
Spolski R, Leonard WJ (2014) Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov 13(5):379–395. doi:10.1038/nrd4296
De Simone V, Franze E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D, Sica GS, Sileri P, MacDonald TT, Pallone F, Monteleone G, Stolfi C (2015) Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 34(27):3493–3503. doi:10.1038/onc.2014.286
Ngiow SF, Smyth MJ, Teng MW (2010) Does IL-17 suppress tumor growth? Blood 115(12):2554–2555; author reply 2556–2557. doi:10.1182/blood-2009-11-254607
Hayata K, Iwahashi M, Ojima T, Katsuda M, Iida T, Nakamori M, Ueda K, Nakamura M, Miyazawa M, Tsuji T, Yamaue H (2013) Inhibition of IL-17A in tumor microenvironment augments cytotoxicity of tumor-infiltrating lymphocytes in tumor-bearing mice. PLoS ONE 8(1):e53131. doi:10.1371/journal.pone.0053131
Gounari F, Chang R, Cowan J, Guo Z, Dose M, Gounaris E, Khazaie K (2005) Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat Immunol 6(8):800–809. doi:10.1038/ni1228
Moser AR, Pitot HC, Dove WF (1990) A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247(4940):322–324. doi:10.1126/science.2296722
Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, Lee DM, Zhang G, Glickman JN, Shin K, Rao VP, Poutahidis T, Weissleder R, McNagny KM, Khazaie K (2007) Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci USA 104(50):19977–19982. doi:10.1073/pnas.0704620104
Sinnamon MJ, Carter KJ, Sims LP, Lafleur B, Fingleton B, Matrisian LM (2008) A protective role of mast cells in intestinal tumorigenesis. Carcinogenesis 29(4):880–886. doi:10.1093/carcin/bgn040
Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK, Regev A (2013) Dynamic regulatory network controlling TH17 cell differentiation. Nature 496(7446):461–468. doi:10.1038/nature11981
Bermejo DA, Jackson SW, Gorosito-Serran M, Acosta-Rodriguez EV, Amezcua-Vesely MC, Sather BD, Singh AK, Khim S, Mucci J, Liggitt D, Campetella O, Oukka M, Gruppi A, Rawlings DJ (2013) Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORgammat and Ahr that leads to IL-17 production by activated B cells. Nat Immunol 14(5):514–522. doi:10.1038/ni.2569
Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR (2000) Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288(5475):2369–2373. doi:10.1126/science.288.5475.2369
Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464(7293):1371–1375. doi:10.1038/nature08949
Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR (2004) An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol 5(1):64–73. doi:10.1038/ni1022
Shibata K, Yamada H, Sato T, Dejima T, Nakamura M, Ikawa T, Hara H, Yamasaki S, Kageyama R, Iwakura Y, Kawamoto H, Toh H, Yoshikai Y (2011) Notch–Hes1 pathway is required for the development of IL-17-producing gammadelta T cells. Blood 118(3):586–593. doi:10.1182/blood-2011-02-334995
Michel ML, Mendes-da-Cruz D, Keller AC, Lochner M, Schneider E, Dy M, Eberl G, Leite-de-Moraes MC (2008) Critical role of ROR-gammat in a new thymic pathway leading to IL-17-producing invariant NKT cell differentiation. Proc Natl Acad Sci USA 105(50):19845–19850. doi:10.1073/pnas.0806472105
Obermajer N, Popp FC, Soeder Y, Haarer J, Geissler EK, Schlitt HJ, Dahlke MH (2014) Conversion of Th17 into IL-17A(neg) regulatory T cells: a novel mechanism in prolonged allograft survival promoted by mesenchymal stem cell-supported minimized immunosuppressive therapy. J Immunol 193(10):4988–4999. doi:10.4049/jimmunol.1401776
Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, Becher B, Littman DR, Neurath MF (2009) RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136(1):257–267. doi:10.1053/j.gastro.2008.10.018
Acknowledgments
This work was supported by a United States Department of Veteran Affairs Office of Research and Development Career Development Award-2 (Jason S. Gold).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Mia Shapiro and Bisweswar Nandi: Co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shapiro, M., Nandi, B., Pai, C. et al. Deficiency of IL-17A, but not the prototypical Th17 transcription factor RORγt, decreases murine spontaneous intestinal tumorigenesis. Cancer Immunol Immunother 65, 13–24 (2016). https://doi.org/10.1007/s00262-015-1769-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-015-1769-2