
Abstract
We have demonstrated that immunostimulatory therapies such as interleukin-2 (IL-2) and anti-CD40 (αCD40) can be combined to deliver synergistic anti-tumor effects. While this strategy has shown success, efficacy varies depending on a number of factors including tumor type and severe toxicities can be seen. We sought to determine whether blockade of negative regulators such as cytotoxic T lymphocyte antigen-4 (CTLA-4) could simultaneously prolong CD8+ T cell responses and augment T cell anti-tumor effects. We devised a regimen in which anti-CTLA-4 was administered late so as to delay contraction and minimize toxicities. This late administration both enhanced and prolonged CD8 T cell activation without the need for additional IL-2. The quality of the T cell response was improved with increased frequency of effector/effector memory phenotype cells along with improved lytic ability and bystander expansion. This enhanced CD8 response translated to improved anti-tumor responses both at the primary and metastatic sites. Importantly, toxicities were not exacerbated with combination. This study provides a platform for rational design of immunotherapy combinations to maximize anti-tumor immunity while minimizing toxicities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recently touted as a “Breakthrough Therapy of the Year” for 2013, cancer immunotherapy has gained considerable traction toward becoming a viable alternative to more traditional cytotoxic treatments including radiotherapy and chemotherapy [1]. Despite recent success with treatments such as Provenge®, PD-1 (programmed death-1)/PD-L1 (programmed death ligand 1) blockade, and chimeric antigen receptor (CAR) T cells, the efficacy of many immunotherapeutic regimens has been limited in the clinic by systemic toxicities, best exemplified by interleukin (IL)-2 and interferon (IFN)-α administration. Treatment with FDA-approved immunostimulatory cytokines IL-2 [2, 3] and IFNα [4, 5] has resulted in the need for intensive patient care, and adverse reactions have included sepsis-like syndromes from massive cytokine release, capillary leak syndromes from the induction of high levels of TNFα, neurotoxicity, tachycardia, renal and hepatic dysfunction, and pulmonary edema. Phase I/II clinical studies involving cytokines including IL-12, IL-18, and IL-21 have resulted in similar dose-limiting toxicities requiring maximum tolerated dose (MTD) to be significantly less than that at which tumor efficacy was observed in preclinical regimens [6–8]. Monoclonal antibodies, such as humanized anti-HER2 (trastuzumab) in the treatment of metastatic breast cancer, have resulted in severe cardiotoxicity culminating in heart failure [9]. Therefore, regimens that may maximize or prolong immune activation without repeated administration of strong stimulation may prove advantageous in minimizing toxicities while maximizing anti-tumor efficacy.
CTLA-4 (cytotoxic T lymphocyte antigen 4) is an important immune checkpoint molecule that is involved in T cell contraction following activation and well as regulatory T cell (TREG) function. Upon activation, T cells upregulate CTLA-4, which competes with the co-stimulatory molecule CD28 for binding with CD80/CD86 on antigen-presenting cells [10]. The inhibitory signals transmitted by CTLA-4 contribute to T cell contraction following activation as well as maintaining tolerance to self-antigens [11, 12]. Clinically, CTLA-4 blockade (ipilimumab) has been approved by the FDA in advanced metastatic melanoma [13] and is currently in trials for a number of different malignancies. Consistent with its role in tolerance, CTLA-4 blockade has also led to severe immune-related adverse events (irAEs) in patients undergoing therapy, often times in organs unrelated to the tumor itself [14]. Previously, our laboratory has demonstrated that combination of agonistic anti-CD40 with IL-2 (αCD40/IL-2) results in synergistic anti-tumor responses against a variety of tumor types in young mice [15]. We have shown that treatment with αCD40/IL-2 expands and activates dendritic cell and T cell populations, and anti-tumor efficacy is both CD8 T cell and IFNγ-dependent [15]. Despite success in a number of preclinical tumor models, administration of this regimen is limited due to induction of toxicities with increasing age [16, 17] and/or body fat content as well as following repeated dosing (manuscript in preparation).
In the current study, we compared therapeutic efficacy across different tumors. Analysis of tumor growth curves suggested that tumor regression (as opposed to static or progressive growth) during active therapy is an important indicator of complete response. As the regimen we apply is relatively short (11 days), we hypothesized that prolonged immunotherapy may lead to improved efficacy. While repeated dosing of CD40/IL-2 to prolong immune activation enhanced toxicities leading to mortality, we show that combination of a single course of anti-CD40 and IL-2 combined with subsequent immune checkpoint blockade using anti-CTLA-4 can enhance and prolong the activation of CD8 T cells. CD8 T cell frequency and function were enhanced at the peak of treatment and maintained for the duration of CTLA-4 administration. For the first time, we also illustrate that CTLA-4 may also play a role not only in the contraction of antigen-specific T cells but bystander-activated CD8 T cells as well. This enhanced T cell activation resulted in improved systemic anti-tumor effects, with decreased growth of the primary tumor, decreased metastases, and enhanced survival without the manifestation of gross toxicities.
Materials and methods
Mice
C57BL/6 and congenic Ly5.1 C57BL/6 mice were purchased from the Animal Production Area of the National Cancer Institute (Frederick, MD), or Jackson Laboratories (Bar Harbor, ME). Female OT-I (C57BL/6-Tg(TcraTcrb)1100Mjb/J) and C57BL/6 mice and wild-type controls were purchased from the Jackson Laboratory (Bar Harbor, ME) and were used at 8–24 weeks of age. All studies were performed with the consent and approval of the University of California, Davis Institutional Animal Care and Use Committee, protocol number 15359.
Immunotherapeutic treatment
C57BL/6 and BALB/c mice were treated with agonistic rat anti-mouse anti-CD40 (clone FGK115B3) (85 or 65 ug i.p. for 5 days, respectively) and recombinant human interleukin-2 (5 × 10^5 IU i.p. twice a week for 2 weeks) as previously described [18]. The anti-mouse CD40 antibody (clone FGK115B3) was generated via ascites production in our laboratory, as previously described [18]. Recombinant human IL-2 (IL-2; TECIN Teceleukin) was provided by the National Cancer Institute repository (Frederick, MD). Blocking antibody to CTLA-4 (clone UC10, a kind gift from Dr. Bruce Blazar) or hamster IgG was administered at 100ug/0.2 ml every other day beginning on day 0 or 7 for up to 6 injections, depending on experimental end point.
Tumor models
The 3LL Lewis lung carcinoma line and the 4T1 mammary carcinoma line were originally purchased from American Type Culture Collection (ATCC, Manassas, VA) and propagated in our facility as previously described [15, 19]. For the 3LL s.c. tumor model, 106 3LL cells were implanted subcutaneously into the shaved flank of 8- to 10-week-old C57BL/6 mice. For the 4T1 orthotopic tumor model, 5 × 104 4T1 cells were injected into the right mammary fat pad of BALB/c mice.
Serum cytokine quantification by cytometric bead array
Serum levels of TNFα, IFNγ, and IL6 were quantified by multiplex measurement using the Becton–Dickinson Cytometric Bead Array CBA (BD Biosciences, San Jose, CA) kit according to manufacturer’s instructions. Each sample was run in triplicate. Upon analysis of raw data, the MFIs in the detection reagent channel of each bead cluster were quantified. Serum cytokine concentrations were then extrapolated relative to a standard curve created by serial dilution of multiplexed mouse standard cytokines run in parallel. Data were acquired on a BD Fortessa cell analyzer using FACSDiva® software (Becton–Dickinson, San Jose, CA) and analyzed using FlowJo software (TreeStar, Ashland, OR). Standard regressions and sample statistical significance were determined using GraphPad Prism data analysis software (GraphPad Software Inc., La Jolla, CA).
Adoptive transfer experiments
The OT-I adoptive transfer experiments were performed as previously described [18]. Briefly, prior to transfer OT-I Tg mice were vaccinated with ovalbumin/incomplete Freund’s emulsion to expand the memory T cell population. Mice were rested, OT-I CD8 T cells were purified from vaccinated mice, and 5 × 106 purified OT–I cells were injected into congenic recipients. Treatments commenced 1 day post-adoptive transfer.
Flow cytometry
Single cell suspensions were labeled with Fc block (purified anti-mouse CD16) (BD Pharmingen, San Diego, CA) and antibodies for 20 min and washed twice with a staining buffer consisting of 5 % FBS (Gemini Bio-products, Sacramento, CA) in DPBS (Mediatech, Herndon, VA). Samples were analyzed using a custom-configured Fortessa using FACSDiva® software (Becton–Dickinson, San Jose, CA). The BD Cytofix/Cytoperm™ kit (Becton–Dickinson) was used for granzyme staining per manufacturer’s instructions. Data were analyzed using FlowJo software (TreeStar, Ashland, OR). Antibodies included: PE-Cy5-conjugated anti-mouse CD62L, APC-Cy7-conjugated anti-mouse CD25 (clone PC61), PE-Cy5- conjugated anti-mouse CD44, PE and PE-Cy7-conjugated anti-mouse NKG2D, PE-conjugated anti-mouse TCR Vα2, PE-conjugated anti-mouse PD-1 (eBioscience, San Diego, CA); FITC-conjugated anti-mouse TCR Vβ5.1/5.2, FITC-conjugated anti-mouse CD4, and APC-Cy7-conjugated CD25 (clone PC61) (BD Pharmingen, San Diego, CA); Alexa Fluor 700-conjugated anti-mouse CD8, Pacific Blue-conjugated anti-mouse CD44 and anti-mouse CD45.2 (BioLegend, San Diego, CA).
Redirected lysis
Redirect lysis assays were performed as previously described [18, 20]. Briefly, Fc receptor-bearing P815 cells (targets) were radiolabeled with Na51CrO4 (NEZ030S, PerkinElmer, Waltham, MA) and anti-CD3e antibody (clone 1452C11, eBioscience, San Diego, CA). P815 target cells were added to serial diluted, CD8-normalized splenocytes (responders) and incubated at 37 °C for 4 h. Specific lysis was calculated as:
Histopathology and grading score
Liver, lung, and whole intestines were collected at indicated time point, flushed and fixed in 10 % paraformaldehyde, embedded in paraffin, cut in sections, and stained with hematoxylin and eosin (H&E). All tissues were prepared and stained at the Histology Consultation Services, Inc in Everson, WA. Images were captured with an Olympus BX4 microscope equipped with a Q-color3 camera and 10× numerical aperture objective lens. Magnification for each captured image is specified for each experiment in the figure legend. Grading of histopathological inflammation was performed using a grading scale from 0 to 4 in a blind fashion by a board-certified pathologist. Grading score criteria of tissue inflammation and necrosis ranged from 0 to 4 with 0 = no inflammation, 1 = minimal/intermediate, 2 = mild, 3 = moderate, and 4 = severe with tissue necrosis. This grading score for liver, lung, and gastrointestinal tract followed the previously published grading systems [21].
Statistics
Statistical analyses were performed using Prism software (GraphPad Software Inc.). For analysis of three or more groups, the nonparametric ANOVA (analysis of variance) test was performed with the Bonferroni’s post-test. Analysis of differences between two normally distributed test groups was performed using the Student’s t test. Data were tested for normality and variance. A p value of <0.05 was considered significant. All experiments were performed at least twice for reproducibility.
Results
Stimulatory immunotherapy results in rapid expansion of effector populations that contract rapidly following cessation of therapy
We have previously shown that combination of agonistic anti-CD40 with high-dose IL-2 (Supp Fig. 1) results in synergistic anti-tumor effects across a variety of tumor models [15, 20, 22]. Administration of this regimen in the subcutaneous 3LL model results in rapid tumor regression and complete protection from tumor mortality (Fig. 1a) while similar treatment of mice bearing the orthotopic breast cancer model 4T1 confers only a short 1-week increase in survival (Fig. 1b). We sought to better understand these differences in efficacy by evaluating tumor growth in the different models. During therapy, established 3LL tumor growth peaked and began to regress prior to the last IL-2 dose (Fig. 1c). In the 4T1 model, of which all mice eventually died, tumor growth, while slightly delayed, remained progressive throughout the regimen (Fig. 1d). Evaluation of tumor growth curves suggested that tumor regression prior to cessation of therapy directly correlated with tumor efficacy.

Systemic IT induces short-lived immune response that contracts following cessation of therapy. Mice (n = 5 mice/group) were implanted with a, c 106 3LL cells s.c. into the right flank or b, d 5 × 104 4T1 cells orthotopically into the mammary fat pad and then subsequently treated with anti-CD40/IL-2. e–h Mice (n = 3 mice/group) were treated with immunotherapy evaluated for (e) splenic CD8 numbers, (f) fold change, (g) lytic ability, and (h) serum was analyzed for cytokine content by cytometric bead array (CBA). Transparent gray rectangles indicate treatment windows. Data are representative of 2–4 independent experiments. Statistical analysis for survival experiments was performed using log-rank test with Welch’s correction. In other studies, two-way ANOVA with Bonferroni’s post-test. *p < 0.05, **p < 0.01, ***p < 0.001
Previous studies have shown that the anti-tumor effects induced by this regimen are mediated by CD8 T cells and cytokines such as interferon (IFN)-γ [15]. To better understand how critical effectors change over time, we monitored their activity during and after treatment. CD8 T cells undergo a rapid expansion phase peaking at day 12 of therapy, which is followed by a sharp contraction through day 18 (Fig. 1e) immediately following cessation of therapy. Interestingly, the CD8 T cells reach equilibrium at a level twice that of controls, and this new “baseline” is maintained >80 days post-treatment (Fig. 1e, f). However, despite the prolonged CD8 T cell elevation, their function, as broadly assessed by redirected lysis, quickly returns to baseline within a week after treatment ends (Fig. 1g). We also assessed systemic cytokine levels over the same time frame, and much like CD8 lytic ability, serum TNFα, IFNγ, and IL-6 contract completely within a week a stopping treatment (Fig. 1h).
Combination of systemic immunostimulatory therapy with late administration of CTLA-4 blockade improves and prolongs the activation of CD8 T cells
Given the rapid contraction kinetics in the functional capacity of CD8 T cells and the diminution of key cytokines shortly after cessation of therapy, we hypothesized that prolongation of T cell activation and systemic cytokine activity may improve anti-tumor effects in more difficult to treat models such as 4T1. We initially tried to give repeated cycles of the 11-day regimen. However, repeated dosing resulted in mortality and multi-organ failure (data not shown) presumably due to prolonged systemic activation similar to that which occurs in our aged models [17]. Another approach to maintaining the protective T cell response would be to impair contraction of this population through immune checkpoint blockade.
Due to its roles in both contraction and TREG function, we hypothesized that combination of anti-CTLA-4 with IT may prolong T cell activation through both delayed contraction as well as regulatory T cell impairment. However, due to the toxicities observed clinically, we sought to limit its administration to the phase of therapy where the contraction occurs. Therefore, we devised a regimen in which IT given as a typical 11-day regimen (Supp Fig. 1) is supplemented with anti-CTLA-4 (clone UC10) beginning on day 7 and continuing through day 18 (Schema depicted in Fig. 2a). We then monitored systemic T cell phenotype and function at the peak (day 11) and 1 week following cessation of therapy (day 18), a time point in which CD8 activation had reverted to baseline following IT alone. Although both CD4 and TREG numbers expanded in both IT alone or in combination with anti-CTLA-4 (IT + UC10) compared to controls, no differences were observed in CD4 T cell or TREG numbers between IT or IT with late anti-CTLA-4 administration groups (Supp Fig. 2a–b). However, there were increases in both total CD8 frequency and numbers when IT was given with anti-CTLA-4 compared to IT alone at both the peak of IT and 1 week after IT was stopped (Fig. 2b, c), with numbers in combination group equilibrating with IT group by day 25 (1 week after the cessation of anti-CTLA-4 administration). To assess whether the expansion could be improved by initiating CTLA-4 block earlier, we performed an experiment directly comparing concurrent administration (beginning at day 0) with late administration (beginning on day 7) and assessing CD8 expansion at the peak of therapy on day 11. Interestingly, there were no significant differences in CD8 expansion between the two groups (Supp Fig. 3a). Therefore, we proceeded with the late administration schema as the supplementary early doses appeared to provide no additive benefit.

Combination of anti-CTLA-4 with systemic IT delays contraction and prolongs the activation of the protective CD8 response. a Schema depicting treatment regimen. Briefly, mice were treated with rIgG or anti-CD40 on days 0–4. PBS or high-dose IL-2 was administered on days 1, 4, 8, 11. Hamster IgG (hmIgG) or anti-CTLA-4 (UC10) was administered every other day beginning on day 7 and ending on day 17. Mice (n = 3-4 mice/group) were treated with control (rIgG/PBS) or IT (40/2) with either hamster IgG (hmIgG) or anti-CTLA-4 (UC10) as per the regimen in 2A and evaluated for T cell phenotype and function on various time points post-therapy. b Percentage and c numbers of CD8+ T cells on days 11, 18, and 25 following treatment. d Representative dot plots of splenic CD8 T cells based on CD62L and CD44 expression. Percentages of various memory CD8 populations on e) day 11 and f day 18 of therapy. g Lytic ability of splenic CD8 T cells on indicated day of treatment. Data are representative of 2–3 independent experiments. Statistical analysis was performed using two-way ANOVA with Bonferroni’s post-test. *p < 0.05, **p < 0.01, ***p < 0.001
Despite the fact that there were no significant changes in systemic Treg numbers following combination, the role of TREG involvement in the CD8 expansion was further assessed by comparing application of IT in combination with TREG depletion and/or anti-CTLA-4. IT + TREG depletion resulted in a similar expansion of CD8 T cells compared with IT + anti-CTLA-4. However, when IT was combined with both TREG depletion and CTLA-4 blockade, there was an additive effect suggesting distinct mechanisms of action (Supp Fig. 4).
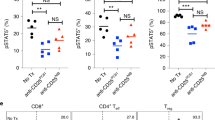
Further evaluation of the phenotype of the CD8 T cells revealed an enrichment in the effector/effector memory population of CD8 T cells, which was apparent more so at the peak of therapy (Fig. 2d, e) but also present at day 18 as well (Fig. 2f). Similar expansions in CD8 T cells and the effector/effector memory phenotype were also found in the lymph nodes (Supp Fig. 5a–b). Finally, analysis of CD8 T cell lytic function by redirected lysis (normalized to account for differences in CD8 frequency) showed, in addition to being expanded, CD8 T cells from IT + UC10-treated mice were more potent effectors than their IT alone counterparts (Fig. 2g). Furthermore, lytic ability was still present at day 18, finally returning to baseline by day 25, whereas IT alone treated mice only showed lytic ability only at day 11 (Fig. 2g). CTLA-4 blockade alone (UC10) had no significant effect on any parameter examined.
Late administration of anti-CTLA-4 results in improved and prolonged expansion of bystander-activated memory CD8 T cells
We have recently shown that the majority of CD8 T cells activated and expanded following immunostimulatory therapies are actually not tumor antigen-specific [20]. In these studies, analysis of activated CD8 T cells revealed that a large population of CD44high (memory/activated) were negative for CD25 (which indicated that TCR had not been engaged), yet displayed elevated NKG2D and granzyme B. Furthermore, intratumoral blockade of NKG2D partially abrogated IT-induced anti-tumor responses implicating bystander-activated (CD25-NKG2D+) memory CD8 T cells as a partial mechanism of anti-tumor immunity following IT [20]. We and others have since observed these cells following influenza infection and established their role in early control of viral replication [18, 23]. The role of CTLA-4 blockade has been extensively studied in T cells following antigen-specific activation [11]. However, a role in bystander-activated memory T cells has yet to be investigated. Since immunostimulatory therapies mainly induce these antigen nonspecific T cells, we next sought to evaluate, for the first time, the effect of CTLA-4 blockade on bystander-activated T cells during treatment with IT. While the proportion of the CD25-NKG2D + bystander-activated T cells of the total CD44highCD8+ T cells was similar between IT and IT + UC10 groups (Fig. 3a,b), the total numbers of bystander-activated CD8 T cells were expanded in the IT + UC10 groups due to the expansion of total CD8+ T cells (Fig. 3c). Furthermore, continued administration of anti-CTLA-4 prolonged activation of bystander-activated cells, which were still present in the IT + UC10 group but not IT alone (Fig. 3a–c) at day 18.

Combination with anti-CTLA-4 improves and prolongs bystander expansion following systemic IT. Mice (n = 3–4 mice/group) were treated with control (rIgG/PBS) or IT (40/2) with either hamster IgG (hmIgG) or anti-CTLA-4 (UC10) as per the regimen in 2A and evaluated for T cell phenotype and function on various time points post-therapy. a Representative histograms of CD44 expression (pregated on CD8) and NKG2D/CD25 expression (pregated on CD44high). b Percentages and c numbers of CD25-NKG2D+ bystander CD8+ T cells. d–f OT-I mice were immunized with OVA/IFA. Thirty days later, OT-I CD8 T cells were isolated and adoptively transferred into WT congenic C57BL/6 mice, which were subsequently treated (n = 2–3 mice/group) as described above. d Schema depicting experimental design. e Representative histograms of NKG2D and CD25 expression on CD45.2 + CD8 + Va2 + CD44high OT-I T cells. f Percentages and g numbers of CD25-NKG2D+ bystander-activated, adoptively transferred OT-I T cells post-treatment. Data are representative of 1–3 independent experiments. Statistical analysis was performed using one- or two-way ANOVA where appropriate with Bonferroni’s post-test. *p < 0.05, **p < 0.01, ***p < 0.001
While the expansion of this phenotype is consistent with our previous studies, it is not sufficient to truly ascertain the nonspecific nature of these cells in bulk T cell populations. OT-I CD8 T cells are specific to chicken ovalbumin (OVA) and therefore represent a T cell that will be nonspecific for our purposes. To further confirm the nonspecific nature of this bystander population, we performed an adoptive transfer of OT-I cells from previously vaccinated OT-I mice (to enrich the memory population) into congenic WT CD45.1 mice. These recipients were subsequently treated and evaluated for expression of the bystander phenotype on adoptively transferred OT-I CD8 T cells (Schema in Fig. 3d). This method accurately depicts the phenotype and expansion of bystander-activated T cells as validated in our previous publications [18, 20]. Consistent with previous studies, frequency (Fig. 3e, f) as well as absolute numbers (Fig. 3g) of bystander phenotype (CD25-NKG2D +) memory OT-I CD8 T cells were significantly increased with IT. As expected, while overall frequency did not change, total numbers of CD25-NKG2D + OT-I CD8 T cells were further expanded when IT was combined with anti-CTLA-4 compared with IT alone (Fig. 3e). Taken together, these data suggest that CTLA-4 blockade compounds the expansion of activated effector CD8 T cells following IT, including bystander-activated CD8 T cells, and can further delay the contraction of these cells after IT is halted.
Improved systemic anti-tumor immunity and tumor CD8 infiltration following combination with anti-CTLA-4 blockade
Due to the improved expansion and function of CD8 T cells following combination therapy, we next evaluated the efficacy of the combination regimen in an orthotopic 4T1 model (Schema in Supp Fig. 1b). Combination of IT with anti-CTLA-4 resulted in delayed tumor growth (Fig. 4a) and enhanced survival (Fig. 4b) compared to IT alone. The 4T1 model is highly metastatic, with tumor detectable in the lymph node, lungs, bone marrow, brain, and liver. Despite the metastatic nature of the 4T1 model, its aggressive growth often results in euthanasia of mice due to primary tumor burden rather than metastatic disease. Therefore, we also evaluated lung metastases by colony assay. While both groups receiving IT had significantly less metastases than control groups, combination of IT with UC10 resulted in significantly less lung metastases than IT alone (Fig. 4c). Finally, we evaluated CD8 T cell expansion in the spleen and infiltration into the tumor. Similar to resting mice, combination with anti-CTLA-4 resulted in expanded CD8 T cells in the spleen as well as improved CD8 infiltration into the tumor (Fig. 4d) and increased lytic ability (Fig. 4e). Importantly, consistent with data in earlier figures showing no difference in T cell proliferation whether CTLA-4 blockade was initiated concurrently or at day 7, no difference in survival was noted between the two conditions as well (Supp Fig. 3b). Taken together, these data confirmed that not only were CD8 T cells more highly activated for an extended period of time, but this enhanced activation state translated into improved anti-tumor immunity.

Combination of anti-CTLA-4 with systemic IT improves anti-tumor responses at primary and metastatic sites. Mice (n = 5–8 mice/group) were implanted with 5 × 104 4T1 orthotopically into the mammary fat pad and treatment was initiated on day 8 post-implantation. Mice were treated with control (rIgG/PBS) or IT (40/2) with either hamster IgG (hmIgG) or anti-CTLA-4 (UC10) as per the regimen in 2A and evaluated for tumor progression and metastases. a Primary tumor growth and b mean survival. c Lung metastases were evaluated by CFU on day 28 post-tumor implantation. d Splenic and intratumoral CD8 T cells were evaluated on day 18 (day 11 of treatment) post-tumor implantation. e Lytic ability of T cells isolated from spleens of treated mice on day 12 as assessed by redirected lysis assay. Data are representative of 1–3 independent experiments. Statistical analysis was performed using one- or two-way ANOVA where appropriate with Bonferroni’s post-test. For survival experiments, log-rank test with Welch’s correction was used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001
Toxicities are not exacerbated by combination with anti-CTLA-4
Clinically, CTLA-4 blockade has resulted in an array of immune-related adverse events (irAEs), some involving tumor-specific autoimmunity and others involving organs unrelated to the tumor, particularly colitis [14]. However, in preclinical studies anti-CTLA-4 alone has not been linked to any significant observable toxicities, and in studies involving combination studies where some toxicities have arisen, these do not mimic what has been seen clinically. In some of the pioneering studies involving anti-CTLA-4 in which it was co-administered in a B16 melanoma model with a GM-CSF-expressing tumor vaccine, significant vitiligo was noted [24]. Since in our studies we were also administering it in combination with another immunotherapy (αCD40/IL-2) which we have also published to exhibit key toxicities under certain scenarios [17, 25], we wanted to be sure that co-administration with anti-CTLA-4 did not further exacerbate toxicities within our model. Therefore, in addition to evaluating immune parameters, we also monitored weight loss, liver enzymes, and organ pathology for any evidence of toxicity. IT alone results in weight loss during the first week of therapy (prior to the time point at which anti-CTLA-4 is initiated), which recovers after the last anti-CD40 injection. Addition of anti-CTLA-4 had no impact on weight loss (Fig. 5a). Serum ALT to measure liver damage follows the same trend in which it is elevated during the first week of therapy and recovers following the last anti-CD40 injection [17]. Similar to weight loss, serum ALT, although significantly elevated in both IT and IT + UC10 groups, was not affected by combination with anti-CTLA-4 (Fig. 5b). Despite the statistical significance, the biological impact of this change is likely insignificant as tenfold to 20-fold increases are typically observed on day two of the regimen (prior to administration of anti-CTLA-4) [17] in young mice and are still typically well tolerated.

Combination of IT with CTLA-4 blockade does not negatively impact toxicities. Mice (n = 3–4 mice/group) were treated with control (rIgG/PBS) or IT (40/2) with either hamster IgG (hmIgG) or anti-CTLA-4 (UC10) as per the regimen in 2A and evaluated for a weight loss and b serum ALT in treatment groups. c–d Hematoxylin and eosin stained c liver (2×) and d gut (4×) sections from all treatment groups were evaluated based on criteria described in detail in Methods section. Quantification of pathology scores from e day 11 and f day 18 is shown. For liver and gut sections, scale bars indicate 2 mm and 600 μm, respectively. Data are representative of 3 independent experiments. Statistical analysis was performed using one- or two-way ANOVA where appropriate with Bonferroni’s post-test. *p < 0.05, **p < 0.01, ***p < 0.001
Systemic inflammation throughout multiple organs is typically observed by day 11 in mice undergoing IT. We next assessed the impact of combination on histopathology in the liver, gastrointestinal tract, and lungs. By day 11 in the liver, IT alone led to extensive lymphocytic infiltrations into the periportal/perivenule regions (black arrows) as well as some intrahepatic aggregates as well (Fig. 5c, e). Although slightly decreased by day 18, lymphocytic aggregations into the liver and periportal/venule regions remained present (Fig. 5f). Comparable infiltrates were noted in the IT + UC10-treated mice at both time points as well (Fig. 5c, f). In the gastrointestinal tract, IT resulted in acute segmental enteritis with mucosal damage (white asterisks), erosion and some ulcerations present at day 11 (Fig. 5d, e) but largely resolving by day 18 (Fig. 5f). Similar to the liver, mice receiving IT + UC10 displayed comparable histopathology in the gastrointestinal tract to IT alone (Fig. 5d, f). Taken together, these data demonstrate that toxicities are not exacerbated when CTLA-4 blockade is administered in combination with IT.
Discussion
Here, we show that rational combination of systemic immunostimulatory therapy with an immune check point inhibitor targeting CTLA-4 can enhance T cell responses during therapy and prolong activation of T cells after cessation of treatment resulting in improved anti-tumor effects. Immunostimulatory therapies have shown great promise in preclinical models often with poor translatability due to unforeseen, dose-limiting toxicities not otherwise observed in murine models. This study suggests that it may be possible to combine these agents with checkpoint inhibitors to potentially (a) increase T cell activation and/or (b) prolong anti-tumor effects after short, high-dose “pulses” of cytokine therapies. Clinical trials have been attempted combining anti-CTLA-4 with high-dose IL-2 but in the opposite order proposed herein and also with both agents given in pulses as opposed to continuous administration of anti-CTLA-4 [26]. Initially, no protective benefit was observed; however, retrospective analysis of patient samples revealed a slight increase in complete response rate [27]. Currently, a phase IV open-label trial is underway evaluating co-administration of high-dose IL-2 with ipilimumab comparing ordering of therapies in treatment naïve metastatic melanoma patients. It will be important to determine how the results of those studies compare to the results presented herein.
In this study, we used blockade of CTLA-4 to prolong activation of the protective CD8 T cell response after immune stimulation was halted. We found that CTLA-4 blockade enhanced and prolonged the activation of all CD8 T cells, including bystander-activated CD8 T cells. Therefore, this study illustrates for the first time that CTLA-4 may also be involved in the contraction of bystander-activated, memory CD8 T cells. Bystander activation of conventional memory CD8 T cells has been well documented in many infectious disease models [23, 28, 29] and, more recently in our group, during immunostimulatory therapies for cancer [20]. While their function has been the subject of debate, we have shown that they have lytic ability and may mediate recognition of target cells, at least in part, through recognition of stress ligands by NKG2D [18, 20]. High-dose cytokine levels are necessary to initiate the bystander expansion and persistence of memory T cells. Cytokine starvation leads to their rapid contraction as is observed following cessation of immunotherapy (Fig. 1e,f). There has been extensive work performed evaluating CTLA-4 in T cell activation by TCR engagement and its role in contraction [11]; however, studies presented herein suggest that co-inhibitory molecules may also play an important role in the contraction of bystander-activated T cells as well. Upon TCR engagement in memory T cells, CTLA-4 is shuttled from intracellular stores to the surface of T cells where it competes with CD28 for binding of co-stimulatory ligands [10]. This is thought to be a mechanism by which memory T cells can avoid activation in the periphery by low affinity TCR engagements, thereby requiring a higher level of co-stimulation [30]. Bystander killing likely does not require co-stimulation; however, the expression of CTLA-4 may confer a higher threshold for TCR-mediated activation while the cell is in the “bystander state.” Another mechanism by which CTLA-4 has been suggested to facilitate inhibition is through reversal of the “STOP signal” [31]. CTLA-4 can reduce dwell time between T cells and a potential target. It is possible that during the contraction phase, blockade of CTLA-4 allows for increased dwell times and processing of survival signals, which may confer the prolonged survival of this cell type in our model.
Despite clinical evidence that CTLA-4 blockade can lead to gross toxicities when given as a monotherapy, late administration with our immunostimulatory regimen did not result in any exacerbated toxicities (Fig. 5). Toxicities with our regimen are consistent with high-dose IL-2 (although not as severe), with an initial cytokine storm syndrome that can be fatal in aged mice [17, 25]. However, the fact that we tailored our regimen to utilize anti-CTLA-4 to delay contraction and therefore administered it at a late time point avoided any added toxicities that may have occurred as a result of co-administration. Enhanced lymphocytic infiltration into organs is also observed following administration of our IT regimen; however, little necrosis/apoptosis is observed in young mice (except in the tumor). This may be due to the fact that the majority of these T cells that are expanded in the organs are bystander-activated, tissue-resident memory T cells. These bystander-activated memory cells, although able to recognize antigen in an NK-like fashion through recognition of stress ligands, have presumably been through multiple rounds of selection: (1) central tolerance in the thymus and (2) activation by antigen (they are memory cells so presumably they have encountered antigen of some sort), rendering them “safe” to the host from initiating autoimmunity.
Despite the recent success of CAR-based therapies and immune checkpoint blockade monotherapies, immunostimulatory therapies for cancer still represent an important class of therapies for treatment of this disease. Unlike vaccine-based regimens, they favor the potential for the release of novel antigens in an adjuvant setting allowing for the generation of a multifaceted immune response to many different epitopes. This study shows that despite their significant toxicities and modest anti-tumor effects as monotherapies, sequential combination of stimulatory therapies with immune checkpoint inhibitors may result in greater synergy with a fraction of the toxicity. It will be important moving forward to build upon this idea to combine various classes of immunotherapies to complement one another and maximize their strengths while minimizing their toxicities.
Abbreviations
- αCD40:
-
Anti-CD40
- ALT:
-
Alanine aminotransferase
- ANOVA:
-
Analysis of variance
- CAR:
-
Chimeric antigen receptor
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
- FDA:
-
Food and Drug Administration
- IFN:
-
Interferon
- IL:
-
Interleukin
- IT:
-
Immunotherapy with anti-CD40/IL-2
- MTD:
-
Maximum tolerated dose
- NKG2D:
-
Natural killer group 2D
- OVA:
-
Ovalbumin
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed death ligand-1
- TCR:
-
T cell receptor
- TNF:
-
Tumor necrosis factor
- TREG :
-
Regulatory T cell
References
Couzin-Frankel J (2013) Breakthrough of the year 2013. Cancer immunotherapy. Science 342(6165):1432–1433. doi:10.1126/science.342.6165.1432
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17(7):2105–2116
Margolin KA, Rayner AA, Hawkins MJ, Atkins MB, Dutcher JP, Fisher RI, Weiss GR, Doroshow JH, Jaffe HS, Roper M et al (1989) Interleukin-2 and lymphokine-activated killer cell therapy of solid tumors: analysis of toxicity and management guidelines. J Clin Oncol 7(4):486–498
Kirkwood JM, Manola J, Ibrahim J, Sondak V, Ernstoff MS, Rao U, Eastern Cooperative Oncology G (2004) A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin Cancer Res 10(5):1670–1677
Eggermont AM, Suciu S, Santinami M, Testori A, Kruit WH, Marsden J, Punt CJ, Sales F, Gore M, Mackie R, Kusic Z, Dummer R, Hauschild A, Musat E, Spatz A, Keilholz U, Group EM (2008) Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet 372(9633):117–126. doi:10.1016/S0140-6736(08)61033-8
Robertson MJ, Cameron C, Atkins MB, Gordon MS, Lotze MT, Sherman ML, Ritz J (1999) Immunological effects of interleukin 12 administered by bolus intravenous injection to patients with cancer. Clin Cancer Res 5(1):9–16
Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL (1997) Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 90(7):2541–2548
Petrella TM, Tozer R, Belanger K, Savage KJ, Wong R, Smylie M, Kamel-Reid S, Tron V, Chen BE, Hunder NN, Hagerman L, Walsh W, Eisenhauer EA (2012) Interleukin-21 has activity in patients with metastatic melanoma: a phase II study. J Clin Oncol 30(27):3396–3401. doi:10.1200/JCO.2011.40.0655
Feldman AM, Lorell BH, Reis SE (2000) Trastuzumab in the treatment of metastatic breast cancer: anticancer therapy versus cardiotoxicity. Circulation 102(3):272–274
Krummel MF, Allison JP (1995) CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182(2):459–465
Walker LS, Sansom DM (2011) The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol 11(12):852–863. doi:10.1038/nri3108
Khattri R, Auger JA, Griffin MD, Sharpe AH, Bluestone JA (1999) Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J Immunol 162(10):5784–5791
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723. doi:10.1056/NEJMoa1003466
Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE, Kammula US, Topalian SL, Sherry RM, Kleiner D, Quezado M, Lowy I, Yellin M, Rosenberg SA, Yang JC (2006) Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol 24(15):2283–2289. doi:10.1200/JCO.2005.04.5716
Murphy WJ, Welniak L, Back T, Hixon J, Subleski J, Seki N, Wigginton JM, Wilson SE, Blazar BR, Malyguine AM, Sayers TJ, Wiltrout RH (2003) Synergistic anti-tumor responses after administration of agonistic antibodies to CD40 and IL-2: coordination of dendritic and CD8+ cell responses. J Immunol 170(5):2727–2733
Bouchlaka MN, Murphy WJ (2013) Impact of aging in cancer immunotherapy: the importance of using accurate preclinical models. Oncoimmunology 2(12):e27186. doi:10.4161/onci.27186
Bouchlaka MN, Sckisel GD, Chen M, Mirsoian A, Zamora AE, Maverakis E, Wilkins DE, Alderson KL, Hsiao HH, Weiss JM, Monjazeb AM, Hesdorffer C, Ferrucci L, Longo DL, Blazar BR, Wiltrout RH, Redelman D, Taub DD, Murphy WJ (2013) Aging predisposes to acute inflammatory induced pathology after tumor immunotherapy. J Exp Med 210(11):2223–2237. doi:10.1084/jem.20131219
Sckisel GD, Tietze JK, Zamora AE, Hsiao HH, Priest SO, Wilkins DE, Lanier LL, Blazar BR, Baumgarth N, Murphy WJ (2014) Influenza infection results in local expansion of memory CD8(+) T cells with antigen non-specific phenotype and function. Clin Exp Immunol 175(1):79–91. doi:10.1111/cei.12186
Bouchlaka MN, Sckisel GD, Wilkins D, Maverakis E, Monjazeb AM, Fung M, Welniak L, Redelman D, Fuchs A, Evrensel CA, Murphy WJ (2012) Mechanical disruption of tumors by iron particles and magnetic field application results in increased anti-tumor immune responses. PLoS One 7(10):e48049. doi:10.1371/journal.pone.0048049
Tietze JK, Wilkins DE, Sckisel GD, Bouchlaka MN, Alderson KL, Weiss JM, Ames E, Bruhn KW, Craft N, Wiltrout RH, Longo DL, Lanier LL, Blazar BR, Redelman D, Murphy WJ (2012) Delineation of antigen-specific and antigen-nonspecific CD8(+) memory T-cell responses after cytokine-based cancer immunotherapy. Blood 119(13):3073–3083. doi:10.1182/blood-2011-07-369736
Yousem SA, Berry GJ, Cagle PT, Chamberlain D, Husain AN, Hruban RH, Marchevsky A, Ohori NP, Ritter J, Stewart S, Tazelaar HD (1996) Revision of the 1990 working formulation for the classification of pulmonary allograft rejection: lung Rejection Study Group. J Heart Lung Transplant 15(1 Pt 1):1–15
Berner V, Liu H, Zhou Q, Alderson KL, Sun K, Weiss JM, Back TC, Longo DL, Blazar BR, Wiltrout RH, Welniak LA, Redelman D, Murphy WJ (2007) IFN-gamma mediates CD4+ T-cell loss and impairs secondary antitumor responses after successful initial immunotherapy. Nat Med 13(3):354–360. doi:10.1038/nm1554
Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, Bevan MJ, Zehn D, Prlic M (2013) Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep 3(3):701–708. doi:10.1016/j.celrep.2013.02.020
van Elsas A, Sutmuller RP, Hurwitz AA, Ziskin J, Villasenor J, Medema JP, Overwijk WW, Restifo NP, Melief CJ, Offringa R, Allison JP (2001) Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J Exp Med 194(4):481–489
Mirsoian A, Bouchlaka MN, Sckisel GD, Chen M, Pai CC, Maverakis E, Spencer RG, Fishbein KW, Siddiqui S, Monjazeb AM, Martin B, Maudsley S, Hesdorffer C, Ferrucci L, Longo DL, Blazar BR, Wiltrout RH, Taub DD, Murphy WJ (2014) Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J Exp Med 211(12):2373–2383. doi:10.1084/jem.20140116
Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Haworth LR, Levy C, Kleiner D, Mavroukakis SA, Yellin M, Rosenberg SA (2005) Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol 12(12):1005–1016. doi:10.1245/ASO.2005.03.536
Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, Levy CL, Rosenberg SA, Phan GQ (2012) CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res 18(7):2039–2047. doi:10.1158/1078-0432.CCR-11-1823
Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, Staprans SI, Feinberg MB (2003) Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 18(3):441–452
Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ (2001) Bystander activation of CD8+ T cells contributes to the rapid production of IFN-gamma in response to bacterial pathogens. J Immunol 166(2):1097–1105
Metz DP, Farber DL, Taylor T, Bottomly K (1998) Differential role of CTLA-4 in regulation of resting memory versus naive CD4 T cell activation. J Immunol 161(11):5855–5861
Rudd CE (2008) The reverse stop-signal model for CTLA4 function. Nat Rev Immunol 8(2):153–160. doi:10.1038/nri2253
Acknowledgments
We would like to acknowledge both Monja Dawson-Metcalf and Weihong Ma for their excellent technical assistance. We would also like to acknowledge A. A. Hurwitz for helpful discussions and guidance with the preparation of this manuscript. This work was funded by a Grant from the NIH R01 CA095572.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing financial interests to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sckisel, G.D., Mirsoian, A., Bouchlaka, M.N. et al. Late administration of murine CTLA-4 blockade prolongs CD8-mediated anti-tumor effects following stimulatory cancer immunotherapy. Cancer Immunol Immunother 64, 1541–1552 (2015). https://doi.org/10.1007/s00262-015-1759-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-015-1759-4