
Abstract
Although cancer immunotherapy shows efficacy with adoptive T cell therapy (ACT) and antibody-based immune checkpoint blockade, efficacious therapeutic vaccination of cancer patients with tumor-associated antigens (TAAs) remains largely unmet. Current cancer vaccines utilize nonmutated shared TAAs that may have suboptimal immunogenicity. Experimental evidence underscores the strong immunogenicity of unique TAAs derived from somatically mutated cancer proteins, whose massive characterization has been precluded until recently by technical limitations. The development of cost-effective, high-throughput DNA sequencing approaches makes now possible the rapid identification of all the somatic mutations contained in a cancer cell genome. This method, combined with robust bioinformatics platforms for T cell epitope prediction and established reverse immunology approaches, provides us with an integrated strategy to identify patient-specific unique TAAs in a relatively short time, compatible with their potential use in the clinic. Hence, it is now for the first time possible to quantitatively define the patient’s unique tumor antigenome and exploit it for vaccination, possibly in combination with ACT and/or immune checkpoint blockade to further increase immunotherapy efficacy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunotherapy of cancer is based on the evidence that neoplastic cells express proteins quantitatively and/or qualitatively different from normal tissues that can be recognized as TAAs by the host’s immune system [1]. TAAs are processed to generate peptides that are displayed on the surface of cancer cells bound into MHC molecules. Tumor MHC–peptide complexes are recognized by specific T lymphocytes of patients, resulting in antitumor responses that can be enhanced by vaccination with the appropriate TAA [1]. The TAAs currently utilized in clinical trials are nonmutated proteins/peptides shared by different cancer types, which generate vaccines that can be conveniently administered to different patients. Nonmutated shared TAAs belong to three major groups: (1) cancer–germline antigens, not or weakly expressed by normal adult tissues and re-expressed by cancer cells; (2) overexpressed antigens, expressed by some normal tissue but up regulated in cancer; and (3) differentiation antigens, expressed by both the normal tissue from which the tumor derives, such as melanocytes and melanoma or prostate epithelium and prostate adenocarcinoma [2, 3]. Immunization of patients with nonmutated shared TAAs often induces specific immune responses that can be increased by new molecularly defined adjuvants [4–6]. However, despite the fact that the first dendritic cell-based vaccine for prostate cancer (Provenge®) [7] was recently approved for the USA and Europe, they have produced clinical responses in only 5–10 % of the treated patients [1, 5]. The poor immunogenicity of nonmutated shared TAAs is likely owed to multiple mechanisms. These TAAs are self-antigens; therefore, to prevent autoimmunity, high affinity T cells are deleted in the thymus by mechanisms of central tolerance that shape the repertoire of specific T lymphocytes, resulting in the export of the low affinity clones [1, 2, 8, 9]. Moreover, shared nonmutated TAAs trigger a series of peripheral mechanisms of tolerance that may restrain the antitumor T cell response induced by immunization, resulting in a poor clinical effect [8–11]. Efforts are therefore required to identify more immunogenic TAAs to induce clinically efficacious antitumor immune responses.
The fourth group of TAAs includes those deriving by DNA somatic mutations or chromosomal aberrations occurring during neoplastic transformation and tumor progression. These so-called tumor-unique antigens were the first to be discovered in animal models and represent truly tumor-specific, strongly immunogenic antigens eliciting cancer rejection by the host [1, 2, 12–15]. Their existence was inferred first in classic experiments in which mice rejected a first challenge with one tumor and then a second challenge with the same tumor, but not with independent tumors of the same histotype [12–14, 16]. The presence of tumor-specific unique peptides associated with heat shock proteins hsp70, hsp90, and gp96 has also been implicated by studies showing tumor-specific immune responses elicited by tumor-derived HSP–peptide complex vaccines extracted form mouse or human tumors [17–22]. Their identification has proceeded essentially via labor-intensive, low-throughput cellular approaches based on the use of small numbers of tumor-specific T cells clones as cellular probes to screen peptides or cDNA library pools, obtained from autologous cancer cells [1, 14, 15, 23, 24]. These serious technical limitations have precluded until recently the systemic molecular characterization of the unique mutated TAAs, thus preventing their exploitation in cancer vaccines.
Massive identification of somatic mutations in cancer cell genes
Random mutagenesis throughout the genome is the hallmark of neoplastic transformation and occurs by nucleotide substitutions, deletions, insertions, or gross chromosomal events [25–28]. The new massive parallel DNA sequencing technologies have rapidly evolved to presently provide a rather cost–effective tool to identify at once all the mutations contained in the exomes of cancer cells [28–31]. About 95 % of the cancer gene somatic mutations are single-base substitutions, whereas the remaining are deletions or insertions of one or a few bases. About 90 % of the base substitutions result in missense changes that alter the protein sequence [26–28]. Common solid cancers, such as those arising from colon, breast, brain, or pancreas harbor on average 33–66 somatic mutations [26–28]. Other tumors display either higher mutational rate than average, typically melanomas, and lung tumors that contain about 200 nonsynonymous mutations per tumor, or less frequent mutations, such as pediatric tumors and leukemia’s that harbor on average 9.6 point mutations [26–28]. The number of mutations directly reflects the different involvement of mutagens, such as cigarette smoking in lung cancer or UV light in melanoma, in the pathogenesis of each tumor type [26–28]. Defects in DNA repair impact also in the accumulation of cancer gene mutations. For example, microsatellite instable (MSI) colorectal cancers with mismatch repair defects harbor thousands of mutations, higher than their microsatellite stable (MSS) counterparts or even than lung tumors and melanomas [26–28, 32]. The genes that harbor somatic mutations at a statistically significant rate or pattern in the same tumor type, compared to the healthy tissue counterpart, are defined candidate cancer genes (CAN-genes) [26–28]. The mutations that confer a selective growth advantage to the tumor cell are called “driver” mutations, whereas the “passenger” mutations do not appear to confer selective growth advantage [26–28].
T cell response against unique versus self-TAAs
Cancer gene somatic mutations generate many tumor-specific proteins bearing amino acid substitutions, which frequently differ from tumor to tumor, therefore forming potential neo-antigens for the host’s immune system. Unique mutated TAAs characterized each single mouse tumor [1, 2, 14]; therefore, they represent the only true tumor-rejection Ags not expressed by normal tissue. Being mutated in comparison with self, the unique TAAs are expected to behave like nonself foreign Ag and hence induce high avidity T cell response rather then tolerance [2]. Classic studies in mice revealed that these unique mutated TAAs are indeed immunodominant over shared nonmutated ones and play a crucial role in tumor rejection in vivo, underscoring their stronger immunogenicity for the autologous immune system [33, 34]. Similar studies in human tumors, particularly in melanoma, suggest that the T cell response is also dominated by the recognition of unique rather then nonmutated TAAs [1, 34, 35]. Up to 45 unique somatically mutated Ags (see CancerImmunity.org) have been identified, which are expressed by different human tumor types and capable of generating mutated peptides inducing vigorous autologous T cell responses in vitro and, in melanoma, also in vivo [1, 34, 36]. The molecular characterization of unique TAAs from both mouse and human tumors revealed that they often correspond to mutated proteins involved in neoplastic transformation [2, 14, 36]. This would likely counteract tumor immune escape, explaining the greater efficacy of mutated TAAs in eliciting tumor rejection in comparison with shared TAAs, in animal models, and now also in human melanoma and cholangiocarcinoma [37–39]. However, it has also been shown that T cell recognition of strongly immunogenic unique epitopes derived from mutated proteins, although not clearly related to oncogenesis, resulted in the selection of antigen-loss tumor escape variants in a mouse sarcoma model [40]. Hence, targeting multiple unique strong antigens by T cells may be the way to overcome the problem of single epitope loss.
As anticipated above, the immunogenicity of TAAs may also depend on the activation of peripheral mechanisms of tolerance, such as the induction of CD4+CD25+T regulatory cells (Tregs), a subset of T lymphocytes specialized in the suppression of T effector responses. The suppressive function of this lymphocyte subset is mediated either by direct contact or by releasing suppressive cytokines, such as TGFβ or IL-10 [9, 41]. Tregs mediate peripheral tolerance to self-Ags recognized by low affinity T cells, such as those specific for nonmutated TAAs, in turn impairing their antitumor effects [9]. Conversely, the stronger the TCR signal the more rapidly and completely the responder T cells become refractory to CD4+CD25high suppression [42]. Chemically induced regressor or progressor murine sarcomas, most likely expressing unique mutated TAAs, harbor activated Tregs but the ratio of Tregs to T effector cells critically determines whether the host will reject the tumor [43]. Thus, the stronger response induced by unique mutated TAA may be more successful than that induced by nonmutated self-TAAs in selecting and expanding effector rather than regulatory T cells. The tumor-specific T cells become also negatively regulated by the expression of receptors involved in immune checkpoints pathways, such as CTLA-4 or PD-1 [44]. The blockade of these negative pathways has shown dramatic results in patients with different advanced tumors [45, 46]. Of note, CTLA-4 blockade correlates with the expansion of autologous CD8+ T cells specific for unique epitopes, suggesting that this treatment unleashes frequent T cell responses spontaneously elicited by mutated neo-antigens in patients [38].
An experimental platform for the identification of somatically mutated TAAs
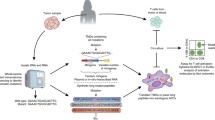
The integration of advanced DNA sequencing techniques, bioinformatics prediction of T cell epitopes and reverse immunology methods provides a platform for the systemic identification of somatically mutated TAAs. First, DNA sequence variants that represent a fraction of a complex sample can be vastly oversampled by massively parallel sequencing, thus enabling statistically significant quantification of low-abundance species, allowing accurate mutation detection in cancer specimens in a manner substantially independent of sample purity (for example, the extent of contaminating stromal DNA) and genomic DNA integrity, unlike conventional “Sanger” sequencing of PCR products [29, 30, 47]. High-throughput whole exome sequencing can be now performed at affordable costs and very high performance with the commercially available instruments [28, 31].
Second, substantial bioinformatics advancements led in recent years to the development of handy algorithms, which can be interrogated with the primary sequence of a given protein to predict the position of possible antigenic epitopes for either CD8+ or CD4+ T cells [48–50]. The algorithms exploit continuously updating databases that identify protein epitopes containing key consensus residues for binding specific HLA class I or class II alleles. Once identified in silico, the antigenicity and immunogenicity of the predicted epitopes are subsequently validated in vitro by using the corresponding synthetic peptides and standard cellular immunology techniques. This whole process, called “reverse immunology,” has led to the identification of T cell epitopes derived from somatically mutated genes, which are recognized by autologous T cells involved in the control of the tumor in a mouse melanoma model [51] or in patients responding to ACT (melanoma and cholangiocarcinoma) [37, 39], or to the treatment with the immune checkpoint blockade Ipilimumab (anti-CTLA4 mAb) (melanoma) [38], or to allogeneic bone marrow transplantation (chronic lymphocytic leukemia) [52]. Whole exome sequencing has also identified immunodominant neoepitopes expressed by chemically induced mouse sarcomas, which elicit potent T cell responses resulting in tumor rejection, but also in cancer immunoediting via T cell-dependent immunoselection of antigen-loss tumor variants [40]. The likelihood to find at least one HLA-A restricted mutated epitope in every mutated CAN-gene found in each patient seems high. Analysis in silico predicts that, for instance, individual colorectal and breast cancers accumulated an average of 7 and 10 unique HLA-A*02:01-restricted epitopes, respectively, corresponding to approximately one new epitope generated for every 10 mutations [53]. Considering that each individual tumor potentially expresses six distinct MHC class I molecules (two alleles each for HLA-A, HLA-B, and HLA-C), the estimated frequency of novel epitopes may be multiplied up to sixfold, suggesting the possibility that individual colorectal and breast cancers can accumulate up to 40 and 60 unique MHC-I restricted epitopes, respectively [53]. We must also further consider the possibility to identify putative CD4+ T cell epitopes from mutated CAN-genes by bioinformatics that will further increase the likelihood of the proposed approach to find tumor-restricted unique antigenic epitopes [39]. The majority of all spontaneously recognized mutated neoepitopes display affinities for HLA-binding comparable to that of their cognate native epitopes. This suggests that somatic mutations preferentially produce neoepitopes that bind with high affinity endogenous TCRs owing to a mutated TCR-contact residue, rather than increasing binding to patients HLA alleles owing to mutated anchor residues [54]. Because the majority of CD8 epitopes are generated by the cytoplasmic cleavage of proteins entering the proteasome degradation pathway, somatic mutations in cancer gene products might also modify their proteasome cleavage, leading to the production of new tumor-restricted CD8 epitopes [1]. While this adds further complexity to the bioinformatics analysis and reverse immunology approach, it does also increase the likelihood that a cancer-related mutation in a given proteins indeed generates new tumor-restricted neo-antigenic epitope.
Unique TAAs for the immunotherapy beyond melanoma: the colorectal cancer model
Most of the information on the mutation-specific T cell response and its role in tumor control were obtained in melanoma. This raises the question as to whether this occurs also in epithelial cancers that comprise over 80 % of all human malignancies. To this respect, it has been recently shown that CD4+ T cells specific for a mutated antigen (erbb2 interacting protein—ERBB2IP) are indeed expanded in TILs and can be harnessed in ACT to mediate regression of a metastatic cholangiocarcinoma [39]. Furthermore, CD8+ T cell response, specific for mutated antigens, was associated with long-term remission following allogeneic hematopoietic stem cell transplantation in chronic lymphocytic leukemia [52]. Colorectal cancer represents a relevant model for investigation, given its frequency, clinical impact, extended molecular characterization, and relevance of the tumor-infiltrating immune response in its prognosis [55, 56]. The availability of sophisticated computational methods in combination with accessible databases and powerful computational infrastructure enables for the first time comprehensive analyses and will pave the way for disentangling tumor and immune heterogeneity [57]. A concept for exploiting these data resources was recently introduced and was used to reconstruct intratumoral immune landscape in colorectal cancer [55]. Using expression profiles from purified immune cells, we could identify cell-type-related gene expression signatures and applied them to microarray data generated from heterogeneous samples from colorectal cancer tumors. The analyses showed highly dynamic intratumoral immune landscapes during tumor progression [55]. This concept can be extended also utilizing data from the Cancer Genome Atlas (TCGA) study (http://cancergenome.nih.gov) [10]. The intratumoral immune landscape, the tumor immunogenicity, and the antigenome in colorectal cancer can be comprehensively characterized using exome-Seq, RNA-Seq, SNP-array, and clinical data. RNA-seq data can be used to assess the type of tumor-infiltrating immune cells and to derive the HLA haplotypes. The binding affinity of the mutated peptides to the corresponding HLA class I allele can be estimated followed by filtering high affinity and expressed neo-antigens. The ploidy and the clonality of mutations can be calculated using SNP-array data, and the molecular phenotypes can be then analyzed with respect to clinical parameters. The results show highly complex immune landscapes and antigenomes of the human colorectal cancer (Trajanoski Z, manuscript submitted). In different experiments, immunogenic somatically mutated epitopes recognized by CD8+ or CD4+ T cells have been identified by re-sequencing the cDNAs encoding the 20 most frequently mutated CAN-genes in different colorectal cancer cell lines and in their cancer stem/initiating cell cultures (Mennonna D and Maccalli C. 2014 in preparation) [58]. The increased frequency of CD8+ T cell precursors specific for a mutated epitopes from Smad4 protein, detected in one patient compared to HLA-matched healthy donors, suggests that the priming of T cell responses specific for the mutated tumor antigens may spontaneously occur in colon cancer patients. These results underscore the efficacy of the approach and highlight the immunogenicity of unique TAAs also in colorectal cancer and their potential for clinical vaccination strategies that can target the most aggressive stem cell component.
Concluding remarks
The characterization of the cancer antigenome is not only important for understanding the mechanisms of tumor–immune cell interaction, but also for developing effective immunotherapies. This information can be used, for example, to stratify patients who would benefit from cancer immunotherapy. Only about 25 % of the patients treated with anti-CTLA-4 antibody respond to therapy [45], and it is of utmost importance to identify patients who bear more immunogenic mutations. Additionally, the identification of the most immunogenic tumor epitopes is a prerequisite for developing personalized cancer vaccines. Tailored vaccine concepts based on the genome-wide discovery of cancer-specific mutations and individualized therapy seem today technically feasible. In this context, the analytical pipelines will be a valuable component for the identification of vaccination targets. However, the number of mutation epitopes that can be included in a vaccine is currently limited to 10–20 due to manufacturing constraints. It is thus critical to pick among the large set of potential epitopes the ones with the highest likelihood of success, i.e., to find an optimal design for the epitope-based vaccine. For this, the development of a framework for selecting the optimal epitope sets based on the available information of a patient is needed.
Abbreviations
- ACT:
-
Adoptive T cell therapy
- Ags:
-
Antigens
- CAN-genes:
-
Candidate cancer genes
- exome-Seq:
-
Exome sequencing
- mAb:
-
Monoclonal antibody
- MSI:
-
Microsatellite instable
- MSS:
-
Microsatellite stable
- RNA-Seq:
-
RNA sequencing
- SNP-array:
-
Single nucleotide polymorphism
- TAAs:
-
Tumor-associated antigens
- TCR:
-
Tell receptor
- TCGA:
-
The Cancer Genome Atlas
- Tregs:
-
CD4+CD25+T regulatory cells
- UV:
-
Ultraviolet
References
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T (2014) Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nature Rev Cancer 14:135–146
Gilboa E (1999) The makings of a tumor rejection antigen. Immunity 11:263–270
Novellino L, Castelli C, Parmiani G (2005) A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother 54:187–207
Parmiani G, Castelli C, Dalerba P, Mortarini R, Rivoltini L, Marincola FM, Anichini A (2002) Cancer immunotherapy with peptide-based vaccines: what have we achieved? Where are we going? J Natl Cancer Inst 94:805–818
Rosenberg SA, Yang JC, Restifo NP (2004) Cancer immunotherapy: moving beyond current vaccines. Nat Med 10:909–915
Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini JC, Romero P (2005) Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 115:739–746
Kantoff PW, Higano CS, Shore ND et al (2010) Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363:411–422
Rivoltini L, Canese P, Huber V et al (2005) Escape strategies and reasons for failure in the interaction between tumour cells and the immune system: how can we tilt the balance towards immune-mediated cancer control? Expert Opin Biol Ther. 5:463–476
Savage PA, Leventhal DS, Malchow S (2014) Shaping the repertoire of tumor-infiltrating effector and regulatory T cells. Immunol Rev 259:245–258
Mortarini R, Piris A, Maurichi A et al (2003) Lack of terminally differentiated tumor-specific CD8+ T cells at tumor site in spite of antitumor immunity to self-antigens in human metastatic melanoma. Cancer Res 63:2535–2545
Hailemichael Y, Dai Z, Jaffarzad N et al (2013) Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med 19:465–472
Prehn RT, Main JM (1957) Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst 18:769–778
Klein G, Sjogren HO, Klein E, Hellstrom KE (1960) Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Res 20:1561–1572
Mumberg D, Wick M, Schreiber H (1996) Unique tumor antigens redefined as mutant tumor-specific antigens. Semin Immunol 8:289–293
Lurquin C, Van Pel A, Mariame B, De Plaen E, Szikora JP, Janssens C, Reddehase MJ, Lejeune J, Boon T (1989) Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell 58:293–303
Foley EJ (1953) Antigenic properties of methylcholanthrene-induced tumors in mice of the strain of origin. Cancer Res 13:835–837
Srivastava P (2002) Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol 20:395–425
Srivastava PK, Duan F (2013) Harnessing the antigenic fingerprint of each individual cancer for immunotherapy of human cancer: genomics shows a new way and its challenges. Cancer Immunol Immunother 62:967–974
Castelli C, Ciupitu AM, Rini F, Rivoltini L, Mazzocchi A, Kiessling R, Parmiani G (2001) Human heat shock protein 70 peptide complexes specifically activate antimelanoma T cells. Cancer Res 61:222–227
Rivoltini L, Castelli C, Carrabba M et al (2003) Human tumor-derived heat shock protein 96 mediates in vitro activation and in vivo expansion of melanoma- and colon carcinoma-specific T cells. J Immunol. 171:3467–3474
Mazzaferro V, Coppa J, Carrabba MG et al (2003) Vaccination with autologous tumor-derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clin Cancer Res 9:3235–3245
Parmiani G, Testori A, Maio M et al (2004) Heat shock proteins and their use as anticancer vaccines. Clin Cancer Res 10:8142–8146
Wolfel T, Hauer M, Schneider J et al (1995) A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269:1281–1284
Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, Boon T (1995) A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci USA 92:7976–7980
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458:719–724
Lawrence MS, Stojanov P, Polak P et al (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218
Lawrence MS, Stojanov P, Mermel CH et al (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505:495–501
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558
Margulies M, Egholm M, Altman WE et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Thomas RK, Baker AC, Debiasi RM et al (2007) High-throughput oncogene mutation profiling in human cancer. Nat Genet 39:347–351
Garraway LA, Lander ES (2013) Lessons from the cancer genome. Cell 153:17–37
Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337
Dudley ME, Roopenian DC (1996) Loss of a unique tumor antigen by cytotoxic T lymphocyte immunoselection from a 3-methylcholanthrene-induced mouse sarcoma reveals secondary unique and shared antigens. J Exp Med 184:441–447
Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wolfel C, Huber C, Wolfel T (2005) The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA 102:16013–16018
Anichini A, Mortarini R, Maccalli C, Squarcina P, Fleischhauer K, Mascheroni L, Parmiani G (1996) Cytotoxic T cells directed to tumor antigens not expressed on normal melanocytes dominate HLA-A2.1-restricted immune repertoire to melanoma. J Immunol. 156:208–217
Parmiani G, De Filippo A, Novellino L, Castelli C (2007) Unique human tumor antigens: immunobiology and use in clinical trials. J Immunol. 178:1975–1979
Robbins PF, Lu YC, El-Gamil M et al (2013) Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19:747–752
van Rooij N, van Buuren MM, Philips D et al (2013) Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 31:439–442
Tran E, Turcotte S, Gros A et al (2014) Cancer immunotherapy based on mutation-specific CD4+ T Cells in a patient with epithelial cancer. Science 344:641–645
Matsushita H, Vesely MD, Koboldt DC et al (2012) Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482:400–404
Thornton AM, Shevach EM (2000) Suppressor effector function of CD4 + CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 164:183–190
von Boehmer H (2005) Mechanisms of suppression by suppressor T cells. Nat Immunol 6:338–344
Bui JD, Uppaluri R, Hsieh CS, Schreiber RD (2006) Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res 66:7301–7309
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nature Rev Cancer. 12:252–264
Hodi FS, O’Day SJ, McDermott DF et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723
Topalian SL, Hodi FS, Brahmer JR et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454
Thomas RK, Nickerson E, Simons JF et al (2006) Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med 12:852–855
Zhang Q, Wang P, Kim Y et al (2008) Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res 36:W513–W518
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M (2008) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res 36:W509–W512
Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, Buus S, Lund O (2008) Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol 4:e1000107
Castle JC, Kreiter S, Diekmann J et al (2012) Exploiting the mutanome for tumor vaccination. Cancer Res 72:1081–1091
Rajasagi M, Shukla SA, Fritsch EF et al (2014) Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 124:453–462
Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP (2008) Epitope landscape in breast and colorectal cancer. Cancer Res 68:889–892
Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ (2014) HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2:522–529
Bindea G, Mlecnik B, Tosolini M et al (2013) Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39:782–795
Galon J, Costes A, Sanchez-Cabo F et al (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Charoentong P, Angelova M, Efremova M, Gallasch R, Hackl H, Galon J, Trajanoski Z (2012) Bioinformatics for cancer immunology and immunotherapy. Cancer Immunol Immunother 61:1885–1903
Volonte A, Di Tomaso T, Spinelli M et al (2014) Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J Immunol. 192:523–532
Acknowledgments
Supported by Associazione Italiana per la Ricerca sul Cancro-AIRC to G. Casorati, G.Parmiani and P.Dellabona.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
This paper is a Focussed Research Review based on a presentation given at the Eleventh Meeting of the Network Italiano per la Bioterapia dei Tumori (NIBIT) on Cancer Bio-Immunotherapy, held in Siena, Italy, 17th–19th October 2013. It is part of a CII series of Focussed Research Reviews and meeting report.
Rights and permissions
About this article

Cite this article
Trajanoski, Z., Maccalli, C., Mennonna, D. et al. Somatically mutated tumor antigens in the quest for a more efficacious patient-oriented immunotherapy of cancer. Cancer Immunol Immunother 64, 99–104 (2015). https://doi.org/10.1007/s00262-014-1599-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-014-1599-7