
Abstract
Malignant gliomas are heavily infiltrated by immature myeloid cells that mediate immunosuppression. Agonistic CD40 monoclonal antibody (mAb) has been shown to activate myeloid cells and promote antitumor immunity. Our previous study has also demonstrated blockade of cyclooxygenase-2 (COX-2) reduces immunosuppressive myeloid cells, thereby suppressing glioma development in mice. We therefore hypothesized that a combinatory strategy to modulate myeloid cells via two distinct pathways, i.e., CD40/CD40L stimulation and COX-2 blockade, would enhance anti-glioma immunity. We used three different mouse glioma models to evaluate therapeutic effects and underlying mechanisms of a combination regimen with an agonist CD40 mAb and the COX-2 inhibitor celecoxib. Treatment of glioma-bearing mice with the combination therapy significantly prolonged survival compared with either anti-CD40 mAb or celecoxib alone. The combination regimen promoted maturation of CD11b+ cells in both spleen and brain, and enhanced Cxcl10 while suppressing Arg1 in CD11b+Gr-1+ cells in the brain. Anti-glioma activity of the combination regimen was T-cell dependent because depletion of CD4+ and CD8+ cells in vivo abrogated the anti-glioma effects. Furthermore, the combination therapy significantly increased the frequency of CD8+ T-cells, enhanced IFN-γ-production and reduced CD4+CD25+Foxp3+ T regulatory cells in the brain, and induced tumor-antigen-specific T-cell responses in lymph nodes. Our findings suggest that the combination therapy of anti-CD40 mAb with celecoxib enhances anti-glioma activities via promotion of type-1 immunity both in myeloid cells and T-cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas account for approximately 30 % of all primary central nervous system tumors and 80 % of malignant brain tumors [1]. The current standard treatment for patients with malignant gliomas is surgery, followed by chemotherapy and radiation therapy [2]. Despite recent advances in cancer therapy, prognosis is dismal; especially for patients with the most aggressive and common malignant glioma (i.e., glioblastoma), the median survival is less than 15 months [1]. Hence, we seek to develop more effective treatments.
Myeloid cells often infiltrate intensively in cancer [i.e., tumor-associated macrophages (TAMs)]. Similar to Th1/Th2 polarization of T-cells, macrophages are also categorized into 2 groups according to the functional phenotypes: M1 and M2 cells [3]. TAMs are typically characterized as M2-like phenotype, which play a role in promoting tumor growth and suppressing type-1 immunity [3], and the degree of TAM accumulation correlates with a poor prognosis in a variety of cancer types [4]. Myeloid-derived suppressor cells (MDSCs) are a cancer-associated heterogeneous population of immature myeloid cells and also critical mediators of immune suppression and tumor progression [5]. In contrast, M1 macrophages contribute to type-1 immune responses and demonstrate direct tumoricidal activities [3].
CD40 is a member of the tumor necrosis factor (TNF) receptor superfamily and expressed on antigen-presenting cells (APCs), such as macrophages and dendritic cells [6]. Signaling via CD40 on APCs initiates M1 polarization, including production of cytokines such as IL-12, TNF-α and nitric oxide (NO), and up-regulation of costimulatory molecules [7–10]. Agonistic CD40 mAb has been shown to activate myeloid cells, promote their M1 polarization and induce antitumor responses both in mice and humans [11–13].
Recent studies have demonstrated that TAMs frequently up-regulate COX-2 expression, thereby enhancing prostaglandin E2 (PGE2) production [14, 15]. PGE2 is one of the key factors in development and accumulation of MDSCs [16–18]. Inhibition of PGE2 biosynthesis in tumor-bearing mice retards tumor progression and reverts immunosuppressive functions of MDSCs [16, 17], and we have recently reported that COX-2 blockade delays glioma development in mice through reduction of MDSCs [18].
In this study, we investigated whether a combination of an agonistic anti-CD40 mAb combined with the COX-2 inhibitor celecoxib induces potent anti-glioma activity in preclinical glioma models through modulation of two independent pathways related to myeloid cell functions. Our findings demonstrate that the combination therapy enhances anti-glioma effects through activation of both myeloid cells and T-cells.
Materials and methods
Mice
C57BL/6 background wild-type (WT) and B6.129S2-Cd8a tm1Mak/J (CD8-deficient) mice (female, 6–8 week old) were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were maintained and handled in accordance with the Animal Facility at the University of Pittsburgh per an Institutional Animal Care and Use Committee-approved protocol.
Antibodies
Rat anti-mouse CD40 mAb (FGK4.5), rat IgG2a (2A3) and rat IgG2b (LTF-2) isotype controls were purchased from BioXcell (West Lebanon, NH). Rat anti-CD4 mAb (GK1.5) was purchased from Taconic (Hudson, NY). The following antibodies were purchased from BD Biosciences (San Jose, CA): anti-CD80 (16-10A1), anti-CD86 (GL1), anti-CD45 (30-F11), anti-CD11b (M1/70), anti-Gr-1 (RB6-8C5) and anti-CD25 (PC61). The following antibodies were purchased from eBioscience (San Diego, CA): anti-CD3 (145-2C11), anti-CD4 (GK1.5), anti-CD8 (53-6.7), anti-IFN-γ (XMG1.2) and anti-Foxp3 (NRRF-30).
Induction of de novo gliomas by intraventricular transfection of Sleeping Beauty transposon-flanked proto-oncogenes
The procedure has been described previously [19]. Briefly, the following DNA plasmids were mixed with in vivo compatible DNA transfection reagent, in vivo-JetPEI (Polyplus Transfection, New York, NY): pT2/C-Luc//PGK-SB100 (0.06 μg/mouse), Sleeping Beauty transposon (SB)-flanked pT2/CAG-NRasV12 (0.12 μg/mouse) and pT2/shp53/mPDGF (0.12 μg/mouse), and injected into the right lateral ventricle of neonates.
Glioma cells
GL261 cells were kindly provided by Dr. Robert M. Prins (University of California Los Angeles, Los Angeles, CA). Quad-GL261 cells stably expressing human gp10025–33, OVA257–264, OVA323–339 and mouse I-Eα52–68 were generated [20] and kindly provided by Dr. John R. Ohlfest (University of Minnesota, Minneapolis, MN). For establishment of the SB28 glioma cell line, de novo glioma was induced as described above in a neonatal C57BL/6 mouse in our laboratory. The brain tissue was harvested at 7 weeks following the glioma induction, minced and then seeded on 100 mm dish in Dulbecco’s modification of Eagle’s medium [DMEM, Cellgro (#10-013), Mediatech, Inc., Manassas, VA] containing 50 U/ml penicillin, 50 μg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, 100 μM 2-mercaptoethanol and 10 % heat-inactivated fetal bovine serum. The medium was changed once a week until glioma cells grew as monolayers. Subsequently, the cells were subcultured and cloned by limiting dilution. A clone with the highest luciferase activity (i.e., transgene expression) was selected and grown as the SB28 cell line.
Live animal imaging
The bioluminescence imaging (BLI) was performed by Xenogen IVIS200 Imaging System (Caliper, A PerkinElmer, Waltham, MA) after 10 min of intraperitoneal injection of D-Luciferin (Gold Biotechnology, Inc., St. Louis, MO, 4.5 mg in 150 μl of PBS/mouse) under isoflurane anesthesia at the In Vivo Imaging Facility of the University of Pittsburgh Cancer Institute.
Therapeutic studies in mouse models
C57BL/6 mice were intracranially inoculated with Quad-GL261 (1 × 105) or SB28 (5 × 104) cells in 2 μl of PBS at the bregma 3 mm to the right side of sagittal suture and 3.5 mm below the surface of skull using stereotactic frame (David Kopf Instruments, Tujunga, CA), stereotaxic injector (Stoelting Co., Wood Dale, IL) and 10 μl Hamilton syringe (Hamilton, Nero, NV) under anesthesia on day 0. The therapy started on day 13: anti-CD40 mAb (100 μg in 100 μl of PBS/mouse) on day 13 or days 13 and 23 intraperitoneally (i.p.), and/or celecoxib (Celebrex, Pfizer, 150 ppm in powdered diet) through days 13–33 via diet. In de novo glioma model, after establishment of gliomas was confirmed using BLI between days 30 and 40 following the glioma induction, mice received anti-CD40 mAb (100 μg in 100 μl of PBS/mouse) on days T0 and T10 i.p. and celecoxib (150 ppm in powdered diet) through days T0 to T15 via diet (day T0 was defined as the first day of therapy following the confirmation of glioma establishment). Rat IgG2a isotype control and powdered diet without celecoxib were used for mock treatment. Mice were sacrificed when they showed any of following signs: hunchback, seizures, hemiparesis or weight loss of greater than 20 %.
In vivo cell depletion
C57BL/6 mice bearing Quad-GL261 glioma received i.p. injection of anti-CD4 mAb or rat IgG2b isotype control (each 50 μg in 100 μl of PBS/mouse) twice a week from days 12 to 33 after tumor inoculation.
Isolation of brain-infiltrating leukocytes (BILs)
The procedure to isolate brain-infiltrating leukocytes (BILs) using Percoll (GE Healthcare Life Sciences, Pittsburgh, PA) has been described previously by us [21]. BILs from each individual were used for flow cytometry analysis. For cell sorting of CD11b+Gr-1+ and CD11b+Gr-1− cells, BILs were pooled from 3 to 4 mice in each therapy group.
Flow cytometry
Cell surface, intracellular cytokine and Foxp3 were stained with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, PE combined with a cyanine dye (PE-Cy7)- or allophycocyanin (APC)-conjugated mAbs. Intracellular cytokine staining Cytofix/Cytoperm™ Kit and Foxp3/Transcription Factor Staining Buffer Set were purchased from BD Biosciences (San Jose, CA) and eBioscience (San Diego, CA), respectively, and analyses were performed according to the manufacturer’s instructions. The samples were collected, and the data were analyzed using BD Accuri C6 flow cytometer and software (BD Biosciences, San Jose, CA).
Cell sorting, RNA isolation and quantification of gene expression
CD11b+Gr-1+ and CD11b+Gr-1− cells from BILs harvested on day 5 after therapy were sorted by MoFlo (Beckman Coulter, Inc., Brea, CA) at the Cytometry Facility of the University of Pittsburgh Cancer Institute. Total RNA was extracted from the sorted cells using RNeasy Mini Kit (Qiagen Inc., Valencia, CA), reverse-transcribed by qScript™ cDNA SuperMix (Quanta BioSciences, Gaithersburg, MD), and then amplified with PerfeCTa qPCR SuperMix, ROX™ (Quanta BioSciences, Gaithersburg, MD) and each probe according to the manufacturer’s instructions. The following probes were purchased from Applied Biosystems (Life Technologies, Grand Island, NY): Cxcl10 (Mm00445235_m1), Agr1 (Mm00475988_m1) and glyceraldehyde 3-phosphate dehydrogenase (Gapdh, Mm03302249_g1). Quantitative real-time PCR analysis was performed by StepOne™ Real-Time PCR Systems and Software (v2.3) (Life Technologies, Grand Island, NY). Gapdh was used as an internal control and to normalize each mRNA expression level. Relative expression of mRNAs compared with control samples was calculated by the ddCt method.
CTL analysis
In vitro cytotoxicity was conducted using 6h 51Cr-release assay as described previously [21]. In brief, inguinal lymph node cells were harvested at 8 days after therapy and incubated with GL261 cells loaded with or without synthetic peptide at an E:T ratio of 50:1. OVA257-264 (SIINFEKL) and human gp10025-33 (KVPRNQDWL) peptides were synthesized by the University of Pittsburgh Peptide Synthesis Facility.
Statistical analyses
Log-rank test by GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA) was used to determine significant differences in the survival of mice on Kaplan–Meier plots among the groups. Mean values between two groups were compared using Student’s t test.
Results
Combination therapy of an agonistic anti-CD40 mAb and the COX-2 inhibitor celecoxib results in significantly improved survival of glioma-bearing mice
We first determined whether a combination of an agonist anti-CD40 mAb and celecoxib induces effective anti-glioma responses. C57BL/6 mice bearing Quad-GL261 cells were treated with i.p. administration of anti-CD40 mAb on day 13 and/or celecoxib via powdered diet through days 13–33 after tumor inoculation. Rat IgG2a isotype was used as a control for anti-CD40 mAb. The combination of the two agents significantly prolonged survival in comparison with control mice (P = 0.023; Fig. 1a), while monotherapy with anti-CD40 mAb or celecoxib alone failed to improve survival. While the initial experiment used only one dose of anti-CD40 mAb, we then investigated the effect of adding secondary anti-CD40 mAb injection on day 23 using the same mouse model and schedule as previous experiments demonstrated in Fig. 1a, and found that the therapeutic efficacy of the combined treatment, but not anti-CD40 mAb without celecoxib, was enhanced compared with the control group (P = 0.008; Fig. 1b).

Agonistic anti-CD40 mAb in combination with celecoxib significantly prolonged survival. C57BL/6 mice bearing Quad-GL261 glioma received anti-CD40 mAb i.p. on a day 13 or b days 13 and 23, and/or celecoxib via diet through days 13–33. Kaplan–Meier plot illustrates the survival (*P < 0.05, **P < 0.01, Log-rank test). c C57BL/6 mice bearing established de novo gliomas received the combination therapy or mock treatment. Relative change in tumor volume from day -T1 (baseline; on the day before the treatment started) to day T15 is shown for each mouse as a waterfall plot (n = 11 in each group; *P < 0.05, t test). d C57BL/6 mice bearing SB28 glioma received anti-CD40 mAb i.p. on days 13 and 23 and/or celecoxib via diet through day 13–33. Kaplan–Meier plot illustrates the survival (**P < 0.01, Log-rank test)
To further evaluate the anti-glioma effects of the combination regimen in a more clinically relevant setting, we employed de novo gliomas, which were induced by Sleeping Beauty (SB) transposon-mediated intraventricular transfection of the oncogenes, NRas, PDGF and short hairpin P53 in neonatal C57BL/6 mice. These mice received anti-CD40 mAb or control IgG2a on days T0 and T10 and powdered diet with or without celecoxib through days T0 to T15 after we confirmed the establishment of gliomas using BLI. The relative change in tumor volume from day -T1 (baseline) to T15 was determined by BLI. As shown in Fig. 1c, the combination therapy suppressed the growth of de novo gliomas compared with mock treatment (P = 0.024). Moreover, using de novo glioma-derived SB28 cells, we confirmed that the combination regimen conferred significantly extended survival of SB28-bearing mice using the same treatment schedule as used in experiments shown in Fig. 1b (P = 0.003; Fig. 1d). Taken together, these results demonstrate that the combined therapy with anti-CD40 mAb and celecoxib induces potent anti-glioma activity in multiple mouse models of glioma.
The combination therapy induces maturation and M1-type polarization of CD11b+ cells
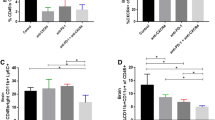
CD40 is a cell-surface molecule expressed on APCs such as dendritic cells and macrophages [6]. To elucidate the effects of the combination therapy on APCs, we evaluated expression levels of maturation markers on CD11b+ cells in splenocytes and BILs on day 2 following the treatment. In splenocytes, anti-CD40 mAb monotherapy and the combination therapy up-regulated CD80 and CD86 (P < 0.001; Fig. 2a) at similar degrees, as well as MHC class I (H-2 Kb) and class II (I-Ab) expression (data not shown). On the other hand, in BILs, the combination therapy, but not anti-CD40 mAb monotherapy, significantly enhanced CD80 expression on CD11b+ cells compared with control and celecoxib treatment (P = 0.021 and P = 0.026, respectively; Fig. 2b). However, none of these treatment groups changed expression levels of CD86 (Fig. 2b), MHC class I or class II in BILs (data not shown).

The combination therapy promoted maturation and M1-like polarization of CD11b+ cells. C57BL/6 mice bearing Quad-GL261 glioma received the combination therapy, monotherapy with anti-CD40 mAb or celecoxib, or mock therapy on day 13 following tumor inoculation. a Spleens and b BILs were harvested on day 15 from individual mice. Expression levels of CD80 and CD86 were evaluated by flow cytometry (n = 5 for control, anti-CD40 and celecoxib groups; n = 4 for the anti-CD40+ celecoxib group; *P < 0.05, ***P < 0.001, t test). c, d Frequencies of CD11b+Gr-1+ and CD11b+Gr-1− cells in the brain were evaluated by flow cytometry on day 5 after therapy (n = 8 for control, n = 6 for monotherapy with anti-CD40 or celecoxib, in each group; n = 7 for anti-CD40+ celecoxib; *P < 0.05, **P < 0.01, ***P < 0.001, t test). e, f BILs were harvested on day 5 posttreatment. CD11b+Gr-1+ and CD11b+Gr-1− BILs were purified from pooled BILs (n = 3–4 in each group) by cell sorting, and mRNA expression levels for Cxcl10 and Arg1 were determined by real-time PCR. Bars and error bars indicate the mean and SD, respectively, from two independent experiments (N.D. not detected; *P < 0.05, **P < 0.01, ***P < 0.001, t test). g Representative flow data of BILs on CD11b+Gr-1+ and CD11b+Gr-1− cells on day 5 posttreatment
We have previously reported that COX-2 blockade suppresses gliomagenesis by inhibiting development and accumulation of CD11b+Gr-1+ immature myeloid cells in the brain using the SB transposon-mediated de novo murine glioma [18]. Hence, we investigated whether anti-CD40 mAb in combination with celecoxib would effectively reduce CD11b+Gr-1+ cells in BILs using the Quad-GL261 glioma model. Unexpectedly, we observed no differences in the frequency of CD11b+Gr-1+ cells among the treatment groups (Fig. 2c, g). However, CD11b+Gr-1+ cells in mice receiving the combination treatment showed significantly higher expression levels of Cxcl10 mRNA compared with those cells in mice receiving control, anti-CD40 mAb or celecoxib alone (P = 0.005, P = 0.003 and P = 0.007, respectively; Fig. 2e). Furthermore, the combination therapy markedly decreased expression of Arg1 mRNA in CD11b+Gr-1+ cells compared with mock and anti-CD40 mAb monotherapy (P < 0.001 and P = 0.041, respectively; Fig. 2f). On the other hand, when we evaluated CD11b+Gr-1− macrophages/microglia in BILs [22], although the percentage of CD11b+Gr-1− BILs was reduced by the combination regimen (Fig. 2d, g), we observed only background levels of Cxcl10 and Arg1 expression in CD11b+Gr-1− BILs isolated from mice with all treatment groups (Fig. 2e, f). Because M1-like macrophages produce CXCL10 [23], which is induced by IFN-γ [24, 25], and arginase-1 expression in mouse macrophages suppresses type-1 immunity [26], these data demonstrate that anti-CD40 mAb in combination with celecoxib reverts the M2-polarized microenvironment of glioma and promotes the M1/type-1 milleau.
T-cells play a major role in the observed anti-glioma activity of the combination regimen
To investigate the possible involvement of T-cells in the observed anti-glioma activity of the combination regimen, we inoculated Quad-GL261 glioma cells in wild-type (WT) and CD8-deficient mice in which CD4+ cells were depleted (T-cell-depleted), and treated them with the combination of anti-CD40 mAb and celecoxib or mock treatment (Fig. 3a). Consistent with Fig. 1b, the combination regimen prolonged the survival of WT mice (P = 0.024). However, in T-cell-depleted mice, the combination treatment did not show improved survival of mice compared to mock treatment (P = 0.578), suggesting that the anti-glioma effect of the regimen is T-cell dependent. We found that the proportion of CD8+ T-cells in BILs significantly increased by anti-CD40 mAb alone or the combination regimen in comparison with control (P = 0.008 and P = 0.026, respectively), whereas the percentage of CD4+ T-cells was not different among the treatment groups (Fig. 3b) on day 5 posttreatment. Similarly, in de novo gliomas, the combination therapy increased the frequency of glioma-infiltrating CD8+ T-cells, but not CD4+ T-cells, compared with mock treatment on day 5 posttreatment (P = 0.034; Fig. 3c).

T-cells were responsible in the observed anti-glioma activity of the combination regimen. a Wild-type (WT) or CD8-deficient mice depleted of CD4+ cells (T-cell-depleted) received intracranial inoculation of Quad-GL261 glioma cells and then received anti-CD40 mAb i.p. on days 13 and 23, and celecoxib via diet through days 13–33. Kaplan–Meier plot illustrates the survival (*P < 0.05, Log-rank test). b C57BL/6 mice bearing Quad-GL261 cells, or c de novo Sleeping Beauty gliomas received anti-CD40 mAb and/or celecoxib. BILs were harvested on day 5 after treatment and analyzed by flow cytometry. Bars and error bars indicate the mean and SD, respectively, from three independent experiments (b n = 8 for control, n = 6 for each of anti-CD40 and celecoxib groups, n = 7 for anti-CD40+ celecoxib, c n = 5 and 6 for control and anti-CD40+ celecoxib, respectively; *P < 0.05, **P < 0.01, t test)
The combination therapy promotes effector functions and enhances antigen-specific cytotoxic activity
To gain better understanding in functional effects of the combined treatment on T-cells, we analyzed IFN-γ production by T-cells and the frequency of CD4+CD25+Foxp3+ regulatory T-cells in BILs derived from Quad-GL261 gliomas on day 5 following the treatment. As demonstrated in Fig. 4a, b, the combination regimen enhanced IFN-γ secretion by CD4+ T-cells and decreased the proportion of CD25+Foxp3+ in CD4+ T-cells in BILs compared to the mock treatment group (P = 0.002 and P = 0.040, respectively). There were no significant changes in IFN-γ production by CD8+ T-cells, although the combination therapy increased the percentage of CD8+ T-cells in BILs. Taken together, these data strongly suggest that anti-CD40 mAb in combination with celecoxib promotes effector functions, but reduces regulatory function of glioma-infiltrating T-cells.

The combination of anti-CD40 mAb and celecoxib improved IFN-γ response, reduced regulatory T-cells and induced antigen-specific cytotoxic activities. C57BL/6 mice bearing day 13 Quad-GL261 glioma received the combination, monotherapy with anti-CD40 mAb or celecoxib, or mock treatment. a IFN-γ production and b Foxp3 expression in BILs were determined by flow cytometry on day 18 (a n = 8 for control, n = 6 for each of anti-CD40 and the combination groups; b n = 5 for control, n = 4 for each of anti-CD40 and the combination groups; *P < 0.05, **P < 0.01, t test). c Inguinal lymph node cells were harvested on day 21, and specific cytotoxicity was evaluated against unpulsed GL261 or GL261 cells loaded with OVA257–264 or gp10025–33 peptide by a 6-h 51Cr-release assay at an E:T ratio of 50:1. Data are presented as mean ± SD. (n = 6 per group; **P < 0.01, t test)
We next evaluated whether the combination regimen would enhance tumor-antigen-specific immune responses. C57BL/6 mice bearing Quad-GL261 glioma, which stably express MHC class I epitopes of OVA and gp100 [20], received the combination or mock treatment on day 13 after tumor inoculation, and inguinal lymph node cells were isolated on day 21 for evaluation of cytotoxic activities. We used GL261 cells loaded with OVA257-264 or gp10025-33 peptide as target cells, as well as non-peptide-loaded GL261 cells as control target cells. As shown in Fig. 4c, the combination therapy enhanced antigen-specific cytotoxic activities against each of OVA257-264 and gp10025-33 peptide epitopes compared with mock treatment (P = 0.004 and P = 0.008, respectively), but we observed no significant cytotoxicity against parental GL261 cells.
Discussion
In this study, we demonstrate that the combined strategy aimed at modulation of myeloid cell functions through two distinct pathways, CD40 signaling and COX-2 blockade, promotes M1-like phenotype of myeloid cells, enhances effector functions, such as IFN-γ production and cytotoxicity, of T-cells and prolongs survival of glioma-bearing mice. Interestingly, the observed efficacy of the combination regimen was dependent on T-cells.
Anti-CD40 mAb directly targets CD40+ tumors, such as B-cell lymphomas, melanomas and carcinomas [27–29]; however, monotherapy with anti-CD40 mAb also shows the potent therapeutic effects in CD40− tumors [28, 30, 31]. There have been mixed observations regarding CD40 expression in gliomas [32, 33]. In our hands, murine Quad-GL261 and SB28 glioma cells do not express detectable CD40 (data not shown). CD40 ligation with an agonistic mAb can trigger both T-cell-dependent and T-cell-independent antitumor responses in mice and patients [34]. While there are several studies with CD40 mAb monotherapy describing the induction of antitumor effects via activation of CD8+ T-cells, NK cells and macrophages [12, 13, 30, 31, 35], we were not able to observe the therapeutic efficacy by anti-CD40 mAb monotherapy in mouse glioma models. Both anti-CD40 mAb monotherapy and its combination with celecoxib enhanced the expression levels of CD80 on CD11b+ BILs and splenocytes at similar levels. However, only splenocytes, but not BILs, demonstrated the up-regulation of CD86, MHC class I and class II (Fig. 2 and data not shown). Although anti-CD40 mAb (with or without celecoxib) significantly increased the proportion of CD8+ T-cells in BILs, only the combination regimen up-regulated IFN-γ production from CD4+ T-cells and reduced the percentage of CD4+CD25+Foxp3+ regulatory T-cells (Figs. 3b, 4b). Taken together, these data suggest that the addition of celecoxib reverses the balance of regulatory T-cells versus helper T-cells toward a more favorable Th1-polarized response.
Blockade of the COX-2 signaling pathway results in reduction of PGE2 biosynthesis, thereby inhibiting inflammation [36], tumor progression and MDSC development in tumor-bearing mice [16–18]. In the current study, however, monotherapy with the COX-2-selective inhibitor celecoxib had no effect on survival and the proportion of CD11b+Gr-1+ cells in BILs (Figs. 1, 2). These contrast with our previous study with de novo mouse gliomas showing that celecoxib delays glioma growth through inhibiting MDSC development and their accumulation in the brain [18]. The main difference between these studies is the selection of mouse glioma models. We think that the effect of celecoxib monotherapy is sufficient to inhibit the growth of de novo gliomas, which are relatively slow growing and poorly immunogenic, but perhaps insufficient for controlling the rapid progression of gliomas established by inoculation of cultured glioma cells in the current study.
CXCL10 is secreted by multiple types of cells including both myeloid cells and lymphocytes in response to IFN-γ and TNF-α [24, 25, 37], and induces chemotaxis, apoptosis, regulation of cell growth and angiogenesis [38, 39]. CXCL10 also plays a critical role in the accumulation of the leukocytes into inflammatory sites [38–40]. Several groups have shown that injection of plasmid vectors expressing CXCL10 inhibits the glioma growth [41, 42], and we have previously demonstrated a critical role of CXCL10 in homing of antigen-specific CD8+ T-cells to brain tumor lesions and survival [18, 21, 43, 44]. Consistent with these previous findings, the combination therapy of anti-CD40 mAb and celecoxib remarkably enhanced Cxcl10 expression in CD11b+Gr-1+ cells (Fig. 2e) as well as CD8+ T-cell infiltration in the brain (Fig. 3b). Although anti-CD40 mAb monotherapy also increased the accumulation of CD8+ T-cells compared with control treatment, it did not induce therapeutic effect. One possible interpretation for these observations is that functions of glioma-infiltrating CD8+ T-cells in mice treated with anti-CD40 mAb alone may be impaired due to high Arg1 expression levels in CD11b+Gr-1+ cells. Arginase I is induced by COX-2 through the PGE2-EP4 receptor signaling and depletes arginine in the tumor microenvironment, thereby suppressing the function of infiltrating T-cells [16, 45]. COX-2 inhibitors, such as celecoxib, block arginase I via inhibition of PGE2 [45]. Hence, the combination of celecoxib may play an important role in the activation of glioma-infiltrating CD8+ T-cells.
GL261 cells express T-cell epitopes derived from melanoma antigens gp100, tyrosinase-related protein 2 (TRP-2), EphA2 and GARC-1 [46–48]. However, in our data, the combination therapy did not induce detectable responses against these endogenously expressed antigen epitopes in parental GL261 cells (Fig. 4c). Hence, to evaluate the induction of adaptive immune responses against tumor-specific antigens, we used Quad-GL261 cells which expressed transgene-derived MHC class I-binding T-cell epitopes from OVA and human gp100 [26]. While we recognize the highly immunogenic nature of these epitopes, we were also able to demonstrate prolonged survival in mice bearing de novo and SB28 gliomas (Fig. 1c, d), which do not express xenogeneic epitopes, indicating that the combination regimen is also effective in weakly immunogenic glioma models.
Although cervical lymph nodes have been shown to be draining lymph nodes in murine gliomas [50], we detected a significant enhancement of tumor-specific T-cell response in the inguinal lymph nodes (Fig. 4c), but not in cervical lymph nodes (data not shown). One possible reason for this observation may be a more profound tumor-induced immunosuppression in draining lymph nodes compared with distal lymph nodes [49] in the GL261 glioma model.
Our findings demonstrate that modulation of two distinct pathways, CD40 signaling and COX-2 blockade, activates not only innate immune response arms, such as myeloid cells, but also T-cell-mediated antitumor functions. Although it may be necessary to extend the duration and/or cycles of treatment regimen to further improve the survival of glioma-bearing mice, the combination therapy induces anti-glioma activity without combining with tumor-antigen-specific immunotherapies, such as antigen-pulsed DC vaccination. Further investigations are warranted to evaluate the addition of antigen-specific vaccines or T-cell adoptive transfer in the current combination regimen to develop more potent immunotherapeutic modalities. Considering the dismal prognosis of malignant glioma, the combination regimen of agonistic anti-CD40 mAb with celecoxib may be a useful strategy for treatment of patients with malignant gliomas.
Abbreviations
- APCs:
-
Antigen-presenting cells
- Arg1:
-
Arginase 1
- BILs:
-
Brain-infiltrating leukocytes
- BLI:
-
Bioluminescence imaging
- Cxcl10:
-
C-X-C motif chemokine 10
- COX-2:
-
Cyclooxygenase-2
- Foxp3:
-
Forkhead box P3
- gp100:
-
Glycoprotein 100
- IFN-γ:
-
Interferon-gamma
- IgG:
-
Immunoglobulin G
- IL:
-
Interleukin
- i.p.:
-
Intraperitoneally
- mAb:
-
Monoclonal antibody
- MDSCs:
-
Myeloid-derived suppressor cells
- MHC:
-
Major histocompatibility complex
- NO:
-
Nitric oxide
- OVA:
-
Ovalbumin
- PGE2:
-
Prostaglandin E2
- SB:
-
Sleeping Beauty
- TAMs:
-
Tumor-associated macrophages
- TNF:
-
Tumor necrosis factor
- WT:
-
Wild-type
References
Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS (2013) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol 15:ii1–ii56
Jovčevska I, Kočevar N, Komel R (2013) Glioma and glioblastoma—how much do we (not) know? Mol Clin Oncol 1:935–941
Allavena P, Mantovani A (2012) Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol 167:195–205
Schmieder A, Michel J, Schonhaar K, Goerdt S, Schledzewski K (2012) Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol 22:289–297
Khaled YS, Ammori BJ, Elkord E (2013) Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol 91:493–502
Grewal IS, Flavell RA (1998) CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol 16:111–135
Kato T, Hakamada R, Yamane H, Nariuchi H (1996) Induction of IL-12 p40 messenger RNA expression and IL-12 production of macrophages via CD40-CD40 ligand interaction. J Immunol 156:3932–3938
Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kämpgen E, Romani N, Schuler G (1996) High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med 184:741–746
Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G (1996) Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med 184:747–752
Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL (2006) Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumour necrosis factor-alpha. Immunology 118:261–270
Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL (2005) CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol 174:6013–6022
Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL (2006) In vivo CD40 ligation can induce T cell-independent antitumor effects that involve macrophages. J Leukoc Biol 79:1181–1192
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH (2011) CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331:1612–1616
Eruslanov E, Kaliberov S, Daurkin I, Kaliberova L, Buchsbaum D, Vieweg J, Kusmartsev S (2009) Altered expression of 15-hydroxyprostaglandin dehydrogenase in tumor-infiltrated CD11b myeloid cells: a mechanism for immune evasion in cancer. J Immunol 182:7548–7557
Eruslanov E, Daurkin I, Vieweg J, Daaka Y, Kusmartsev S (2011) Aberrant PGE2 metabolism in bladder tumor microenvironment promotes immunosuppressive phenotype of tumor-infiltrating myeloid cells. Int Immunopharmacol 11:848–855
Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S (2007) Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 67:4507–4513
Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP (2010) COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 10:464
Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, Ohlfest JR, Okada H (2011) COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res 71:2664–2674
Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, Long C, Demorest ZL, Zamora EA, Low WC, SantaCruz K, Largaespada DA, Ohlfest JR (2009) De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res 69:431–439
Litterman AJ, Zellmer DM, Grinnen KL, Hunt MA, Dudek AZ, Salazar AM, Ohlfest JR (2013) Profound impairment of adaptive immune responses by alkylating chemotherapy. J Immunol 190:6259–6268
Nishimura F, Dusak JE, Eguchi J, Zhu X, Gambotto A, Storkus WJ, Okada H (2006) Adoptive transfer of type 1 CTL mediates effective anti-central nervous system tumor response: critical roles of IFN-inducible protein-10. Cancer Res 66:4478–4487
Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V (1991) Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA 88:7438–7442
Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, Shibata T, Takami T (2008) Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol 83:1136–1144
Luster AD, Unkeless JC, Ravetch JV (1985) Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature 315:672–676
Narumi S, Hamilton TA (1991) Inducible expression of murine IP-10 mRNA varies with the state of macrophage inflammatory activity. J Immunol 146:3038–3044
Munder M (2009) Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol 158:638–651
von Leoprechting A, van der Bruggen P, Pahl HL, Aruffo A, Simon JC (1999) Stimulation of CD40 on immunogenic human malignant melanomas augments their cytotoxic T lymphocyte-mediated lysis and induces apoptosis. Cancer Res 59:1287–1294
French RR, Chan HT, Turr AL, Glennie MJ (1999) CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med 5:548–553
Alexandroff AB, Jackson AM, Paterson T, Haley JL, Ross JA, Longo DL, Murphy WJ, James K, Taub DD (2000) Role for CD40-CD40 ligand interactions in the immune response to solid tumours. Mol Immunol 37:515–526
Todryk SM, Tutt AL, Green MH, Smallwood JA, Halanek N, Dalgleish AG, Glennie MJ (2001) CD40 ligation for immunotherapy of solid tumours. J Immunol Methods 248:139–147
van Mierlo GJ, den Boer AT, Medema JP, van der Voort EI, Fransen MF, Offringa R, Melief CJ, Toes RE (2002) CD40 stimulation leads to effective therapy of CD40(-) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc Natl Acad Sci USA 99:5561–5566
Wischhusen J, Schnider D, Mittelbronn M, Meyermann R, Engelmann H, Jung G, Wiendl H, Weller M (2005) Death receptor-mediated apoptosis in human malignant glioma cells: modulation by the CD40/CD40L system. J Neuroimmunol 162:28–42
Xie F, Shi Q, Wang Q, Ge Y, Chen Y, Zuo J, Gu Y, Deng H, Mao H, Hu Z, Zhou Y, Zhang X (2010) CD40 is a regulator for vascular endothelial growth factor in the tumor microenvironment of glioma. J Neuroimmunol 222:62–69
Vonderheide RH, Bajor DL, Winograd R, Evans RA, Bayne LJ, Beatty GL (2013) CD40 immunotherapy for pancreatic cancer. Cancer Immunol Immunother 62:949–954
Turner JG, Rakhmilevich AL, Burdelya L, Neal Z, Imboden M, Sondel PM, Yu H (2001) Anti-CD40 antibody induces antitumor and antimetastatic effects: the role of NK cells. J Immunol 166:89–94
Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K (1994) Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA 91:3228–3232
Ohmori Y, Hamilton TA (1994) Cell type and stimulus specific regulation of chemokine gene expression. Biochem Biophys Res Commun 198:590–596
Neville LF, Mathiak G, Bagasra O (1997) The immunobiology of interferon-gamma inducible protein 10 kD (IP-10): a novel, pleiotropic member of the C-X-C chemokine superfamily. Cytokine Growth Factor Rev 8:207–219
Liu M, Guo S, Hibbert JM, Jain V, Singh N, Wilson NO, Stiles JK (2011) CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev 22:121–130
Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P (2014) Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev 13:272–280
Enderlin M, Kleinmann EV, Struyf S, Buracchi C, Vecchi A, Kinscherf R, Kiessling F, Paschek S, Sozzani S, Rommelaere J, Cornelis JJ, Van Damme J, Dinsart C (2009) TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther 16:149–160
Jiang XB, Lu XL, Hu P, Liu RE (2009) Improved therapeutic efficacy using vaccination with glioma lysate-pulsed dendritic cells combined with IP-10 in murine glioma. Vaccine 27:6210–6216
Fujita M, Zhu X, Ueda R, Sasaki K, Kohanbash G, Kastenhuber ER, McDonald HA, Gibson GA, Watkins SC, Muthuswamy R, Kalinski P, Okada H (2009) Effective immunotherapy against murine gliomas using type 1 polarizing dendritic cells—significant roles of CXCL10. Cancer Res 69:1587–1595
Fujita M, Zhu X, Sasaki K, Ueda R, Low KL, Pollack IF, Okada H (2008) Inhibition of STAT3 promotes the efficacy of adoptive transfer therapy using type-1 CTLs by modulation of the immunological microenvironment in a murine intracranial glioma. J Immunol 180:2089–2098
Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC (2005) Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med 202:931–939
Prins RM, Odesa SK, Liau LM (2003) Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res 63:8487–8491
Hatano M, Kuwashima N, Tatsumi T, Dusak JE, Nishimura F, Reilly KM, Storkus WJ, Okada H (2004) Vaccination with EphA2-derived T cell-epitopes promotes immunity against both EphA2-expressing and EphA2-negative tumors. J Transl Med 2:40
Iizuka Y, Kojima H, Kobata T, Kawase T, Kawakami Y, Toda M (2006) Identification of a glioma antigen, GARC-1, using cytotoxic T lymphocytes induced by HSV cancer vaccine. Int J Cancer 118:942–949
Ohlfest JR, Andersen BM, Litterman AJ, Xia J, Pennell CA, Swier LE, Salazar AM, Olin MR (2013) Vaccine injection site matters: qualitative and quantitative defects in CD8 T cells primed as a function of proximity to the tumor in a murine glioma model. J Immunol 190:613–620
Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, Rüegg C, Dietrich PY, Walker PR (2005) Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 22:175–184
Acknowledgments
Grant Supports from: The National Institutes of Health (NIH) (2R01 NS055140, 1P01 CA132714) and Musella Foundation for Brain Tumor Research and Information. This project used University of Pittsburgh Cancer Institute (UPCI) shared resources (Animal Facility, In Vivo Imaging Facility and Cytometry Facility) that are supported in part by NIH P30CA047904. The authors thank Dr. Gary Kohanbash and Maki Ikeura (University of Pittsburgh) for their administrative assistance.
Conflict of interest
The authors have no financial conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Akemi Kosaka and Takayuki Ohkuri contributed equally to this work.
Rights and permissions
About this article

Cite this article
Kosaka, A., Ohkuri, T. & Okada, H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother 63, 847–857 (2014). https://doi.org/10.1007/s00262-014-1561-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-014-1561-8