
Abstract
Accumulating evidence suggests that most solid malignancies consist of heterogeneous tumor cells and that a relatively small subpopulation, which shares biological features with stem cells, survives through potentially lethal stresses such as chemotherapy and radiation treatment. Since the survival of this subpopulation of cancer stem cells (CSC) plays a critical role in recurrence, it must be eradicated in order to cure cancer. We previously reported that vaccination with CD133+ murine melanoma cells exhibiting biological CSC features induced CSC-specific effector T cells. These were capable of eradicating CD133+ tumor cells in vivo, thereby curing the parental tumor. In the current study, we indicated that DEAD/H (Asp–Glu–Ala–Asp/His) box polypeptide 3, X-linked (DDX3X) is an immunogenic protein preferentially expressed in CD133+ tumor cells. Vaccination with DDX3X primed specific T cells, resulting in protective and therapeutic antitumor immunity. The DDX3X-primed CD4+ T cells produced CD133+ tumor-specific IFNγ and IL-17 and mediated potent antitumor therapeutic efficacy. DDX3X is expressed in various human cancer cells, including lung, colon, and breast cancer cells. These results suggest that anti-DDX3X immunotherapy is a promising treatment option in efforts to eradicate CSC in the clinical setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There has been increasing evidence that most solid malignancies consist of heterogeneous tumor cells and that a relatively small subpopulation exhibits unique characteristics, including high tumorigenicity, growth as non-adherent spheres, unlimited self-renewal, and differentiation. The members of this unique subpopulation are referred to as cancer stem cells (CSC) because they share biological, biochemical, and molecular features with normal stem cells [1]. All examined cancer cells surviving after cytotoxic chemotherapy and molecular-targeting treatment have been shown to uniformly express CD133, one of the putative CSC markers [2, 3]. Because CSC possess multiple mechanisms to resist cell death—such as an altered chromatin state and an excess of multidrug efflux transporters, anti-apoptotic factors, DNA repair gene products, and stem cell-specific growth signaling—it is likely that cancer can survive as CSC under potentially lethal stresses [2, 4–9]. Unless an effective treatment to eradicate the CSC subpopulation is developed, it will be extremely difficult to achieve a lasting cure.
T cell-mediated immunotherapy, which can in turn mediate antitumor reactivity, provides a potential avenue for developing such a treatment. We previously reported that effector T cells possessed potent antitumor therapeutic efficacy in brain, pulmonary, and skin metastasis models [10, 11]. More recently, we showed that vaccination with CD133+ melanoma cells induced specific CD8+ and CD4+ T cells, including type 17 T helper (Th17) cells and Th1 cells. Moreover, this treatment eradicated CD133+ tumor cells, thereby curing parental melanomas [12]. It is likely that CD133+ melanoma cells possess specific immunogenic antigens and that the antigen-primed T cells mediate antitumor reactivity.
To elucidate the immunogenic proteins that are preferentially expressed in CD133+ tumor cells, we compared protein expression using two-dimensional electrophoresis analyses, identifying 4 proteins. A Mascot search based on mass spectrometry (MS/MS) protein data identified one of those proteins as DEAD/H (Asp–Glu–Ala–Asp/His) box polypeptide 3, X-linked (DDX3X). This protein is a member of the DEAD-box family of ATP-dependent RNA helicases and is located on the X chromosome [13]. DDX3X is evolutionarily well conserved from yeast to humans, suggesting that it is essential for cell survival. Whole-exome analyses revealed that DDX3X is a component of pathogenic β-catenin signaling in medulloblastoma and that mutated DDX3X plays an important role in oncogenesis [14]. DDX3X mutation was discovered in other human malignancies such as chronic lymphocytic leukemia and head and neck squamous cell carcinoma with whole-exome analyses [15, 16].
In this study, we indicated that DDX3X is a major immunogenic target protein of CD133+ melanoma cells. Vaccination with synthesized DDX3X protein exhibited therapeutic efficacy against established skin melanoma, thus curing the tumor. DDX3X is strongly expressed in human cancer cell lines that have CSC markers. These results indicate that anti-DDX3X immunotherapy may be a promising strategy in the efforts to eradicate CSC, thereby curing cancer.
Materials and methods
Mice
Female C57BL/6J (B6) mice were purchased from the CLEA Laboratory (Tokyo, Japan), maintained in a specific pathogen-free environment, and used for experiments at the age of 8–10 weeks. All animal experiments were approved by the Niigata University Ethics Committee.
Tumor cells
B16F10, a melanoma of B6 origin, was maintained in vitro. CD133+ and CD133− tumor cells were isolated using PE-conjugated anti-CD133 mAb (13A4), anti-PE microbeads (Miltenyi Biotec, Auburn, CA, USA) and autoMACS™ (Miltenyi Biotec) according to the manufacturer’s instructions. The cell purity was >90 %.
mAbs and flow cytometry
Hybridomas producing mAbs against murine CD4 (GK1.5, L3T4), CD8 (2.43, Lyt-2), CD3 (2C11), and murine CD62L (MEL14) were obtained from the American Type Culture Collection (Rockville, MD, USA). Anti-CD4 mAb, anti-CD8 mAb, and anti-CD62L mAb were produced as ascites fluid from sublethally irradiated (500 cGy) DBA/2 mice. PE-conjugated anti-CD80 (16-10A), anti-CD86 (GL1), anti-CD62L (MEL14), anti-CD8 (2.43), and anti-CD25 (PC61) mAbs; fluorescein isothiocyanate (FITC)-conjugated anti-Thy1.2 (30-H12); and anti-CD4 (GK1.5) mAbs were purchased from BD PharMingen (San Diego, CA, USA). Analyses of cell-surface phenotypes were conducted through direct immunofluorescent staining of 0.5–1 × 106 cells with conjugated mAbs. In each sample, a total of 10,000 cells were analyzed using a FACScan™ flow microfluorometer (Becton–Dickinson, Sunnyvale, CA, USA). PE-conjugated subclass-matched antibodies used as isotype controls were also purchased from BD PharMingen. The samples were analyzed with CellQuest™ software (BD).
Fractionation of T cells
T cells in the LN cell suspension were concentrated by passage through nylon wool columns (Wako Pure Chemical Industries, Osaka, Japan). To yield highly purified (>90 %) cells with down-regulated CD62L expression (CD62Llow), LN T cells were further isolated by a panning technique using T-25 flasks pre-coated with goat anti-rat immunoglobulin antibody (Ig Ab) (Jackson ImmunoResearch Laboratories, West Grove, PA, USA)/anti-CD62L mAb (MEL14) and sheep anti-rat-Ig Ab/anti-CD62L mAb-coated DynaBeads M-450 (Dynal, Oslo, Norway). In some experiments, cells were further separated into CD4+ and CD8+ cells by depletion using magnetic beads, as described previously [17]. For in vitro experiments, highly purified CD4+ cells were obtained using anti-CD4 mAb-coated Dynabeads and Detachabeads (Invitrogen) according to the manufacturer’s instructions.
Bone marrow-derived DCs
Dendritic cells (DCs) were generated from bone marrow cells (BMs), as described previously. In brief, BMs obtained from the femurs and tibias of treatment-naïve mice were placed in T-75 flasks for 2 h at 37 °C in complete medium (CM) containing 10 ng/mL of recombinant murine granulocyte–macrophage colony-stimulating factor (rmGM-CSF, a gift from KIRIN, Tokyo, Japan). Non-adherent cells were collected by aspirating the medium; they were then transferred into fresh flasks. On day 6, non-adherent cells were harvested by gentle pipetting. CM consisted of RPMI 1640 medium supplemented with 10 % heat-inactivated lipopolysaccharide (LPS) qualified fetal calf serum (FCS), 0.1 mM non-essential amino acids, 1 μM sodium pyruvate, 100 U/mL of penicillin, 100 μg/mL of streptomycin sulfate (all from Life Technologies, Inc.), and 5 × 10−5 M 2-ME (Sigma Chemical Co., St. Louis, MO, USA).
DC/tumor-draining LN cells
BMs and DCs were co-cultured in CM overnight with the same number of irradiated tumor cells (5,000 cGy). B6 mice were inoculated s.c. with 10 × 106 BM-DC and tumor cells in both flanks. Inguinal LNs draining BM-DC and tumor cells were harvested. Single-cell suspensions were prepared mechanically, as described previously [18].
Adoptive immunotherapy
B6 mice were injected s.c. with parental B16-F10 tumor cells in 100 μL of Hanks’ balanced salt solution (HBSS) to establish subcutaneous tumors. Two or three days after the inoculation, the mice were sublethally irradiated (500 cGy) and then infused i.v. with T cells isolated from tumor-draining LNs. These LN cells were stimulated with anti-CD3 mAb (2C11) and cultured in CM containing 40 U/mL of IL-2 for 3 days to obtain a sufficient number of T cells for in vivo experiments, as described previously [11]. The perpendicular diameters of subcutaneous tumors were measured using calipers.
Cytokine ELISAs
T cells were stimulated with immobilized anti-CD3 mAb or tumor antigen-pulsed BM-DCs in CM. Supernatants were harvested and assayed for IFN-γ, IL-4, and IL-17 content by a quantitative “sandwich” enzyme immunoassay using a murine IFN-γ, IL-4, and IL-17 ELISA kit (Genzyme, Cambridge, MA, USA), according to the manufacturer’s instructions.
Immunoblotting assay
Cells were harvested and lysed in Nonidet P-40 buffer containing a protease-inhibitor mixture (Sigma). Equal microgram amounts of proteins were subjected to SDS-7.5 % PAGE and transferred to polyvinylidene difluoride membrane (Millipore). Immunoblots from tumor cells were probed with antibodies against DDX3X (Sigma) and β-actin (Sigma). Secondary antibodies consisted of anti-mouse Ig and anti-rabbit Ig conjugated to horseradish peroxidase (Bio Rad, Dako). Immunoreactive protein bands were visualized using the ECL kit (Pierce). At least, 3 independent experiments were performed for all analyses.
Knockdown of DDX3X by shRNA
Knockdown of DDX3X was obtained using a shRNA lentiviral (pLKO.1-puro) plasmid (Sigma Aldrich). The oligonucleotides containing the DDX3X target sequence that were used were: sequence # 5′-CCGGACGTTCTAAGAGCAGTCGATTCTCGAGAATCGACTGCTCTTAGAACGTTTTTTG. B16 CD133+ cells were added in fresh media, and hexadimethrine bromide (8 μg/mL) was added to each well. The cells were co-transfected with the pLKO.1-puro plasmid plus the packaging vector according to the manufacture’s protocol. The media were changed approximately 16 h after transfection, and the cells were cultured for an additional 48–72 h. Experimental cells were incubated with the fresh media containing puromycin (2.0 μg/mL), and the media were replaced with fresh puromycin-containing media every 3–4 days until resistant colonies could be identified. A minimum of 5 puromycin-resistant colonies were picked, and each clone was expanded for the assay. The efficiency of DDX3X knockdown was determined by immunoblotting.
Statistical analysis
Comparison between groups was performed using Student’s t test. Dynamic tumor-growth data were analyzed by a multivariate, general linear model. Differences were considered significant for P <0.05. Statistical analysis was performed with SPSS statistical software (SPSS, Chicago, IL, USA) or GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Proteome analyses revealed that CD133+ CSCs had specific proteins
We recently reported that immunization with CD133+ tumor cells purified from murine B16 melanoma lines exhibited potent antitumor immunity in vivo [12]. Both CD8+ and CD4+ T cells that were primed by draining LNs with CD133+ tumor cells were found to release cytokines only when stimulated with CD133+ tumor cell antigens, but not when stimulated with CD133− tumor cell antigens. Since our studies suggested that CD133+ melanoma cells possess specific immunogenic antigens, we used two-dimensional electrophoresis to examine whether protein expression patterns differed between CD133+ and CD133− B16 melanoma cells. We found that all proteins except for 7 exhibited the same expression pattern (Fig. 1a). Three proteins (#1 to #3) were preferentially expressed in CD133− tumor cells, whereas 4-protein expression (#4 to #7) was enriched in CD133+ tumor cells (Fig. 1b, c). A Mascot search based on mass spectrometry (MS/MS) protein data identified protein #5 as DDX3X [19].

a Proteome analyses of CD133+ and CD133− B16 melanoma cells. CD133+ or CD133− B16 melanoma cells were washed with phosphate-buffered saline (PBS) and solubilized in lysis buffer (5 M CH4N2O, 2 M CH4N2S, 2 % CHAPS, 2 % SB3-10, 1 % DTT). Protein lysates (70 μg) were separated using two-dimensional protein gel electrophoresis. b Density of #5 plot in CD133− and CD133+ melanoma. c Immunoblotting analysis of DDX3X in CD133− and CD133+ melanoma
DDX3X-specific CD4+ T cells exhibited CD133+ tumor antigen-specific cytokine release
We investigated whether T cells primed with synthesized DDX3X protein could recognize CD133+ melanoma cells. To test this, T cells with down-regulated expression of CD62L (CD62Llow) were isolated as antigen-primed T cells from LNs draining DC that were pulsed with synthesized DDX3X protein (DDX3X/DC). DDX3X-specific CD8+ T cells responded to both CD133− and CD133+ melanoma cells, but not to irrelevant tumor cells that did not express DDX3X (Fig. 2a). In contrast, we found that DDX3X-specific CD4+ T cells secreted IFNγ in a CD133+ melanoma-specific manner and secreted a significantly increased amount of IL-17 on CD133+ tumor antigen stimulation (Fig. 2b, c).

Interferon (IFN)-γ and interleukin (IL)-17A secreted by lymph node (LN) T cells upon DDX3X, CD133−, or CD133+ tumor antigen stimulation. In a 96-well plate, 1 × 105 CD62Llow CD4+ or CD8+ T cells isolated from LNs were stimulated with 1 × 104 DCs in 200 μL complete medium (CM) for 48 h. Stimulator DCs were pulsed overnight with an equal number of 5,000 cGy-irradiated CD133+ or CD133− tumor cells or synthesized DDX3X protein (5 μg/mL) and were purified with CD11c microbeads prior to co-culture. *P < 0.05 and **P < 0.01. a IFNγ secretion by DDX3X-draining CD8+ T cells upon DDX3X, MCA 205, CD133− B16, or CD133+ B16 tumor antigen stimulation. MCA 205 tumor cells are methylcholanthrene-induced murine fibrosarcoma cells and do not express DDX3X. b IFNγ secretion by DDX3X-draining CD4+ LN T cells on DDX3X antigen stimulation. c IFNγ and IL-17 secretion by DDX3X-draining CD4+ LN T cells on CD133+ or CD133− B16 tumor antigen stimulation. d IFNγ secretion by CD133−– or CD133+ B16 melanoma cell–draining CD8+ LN T cells on DDX3X, CD133−, or CD133+ B16 tumor antigen stimulation. e IFNγ and IL-17 secretion by CD133−– or CD133+ B16 melanoma cell–draining CD4+ LN T cells on DDX3X, CD133−, or CD133+ B16 tumor antigen stimulation
Next, we tested whether CD133+ melanoma cell-specific T cells recognized DDX3X and produced cytokines. Consistent with our previous report, we found that CD4+ and CD8+ T cells primed with vaccinated CD133+ melanoma cells produced cytokines in a CD133+ melanoma cell-specific manner (Fig. 2d, e). Although CD133+ melanoma cell-specific CD8+ T cells failed to respond to DDX3X stimulation, CD133+ melanoma cell-specific CD4+ T cells did secrete IFNγ and IL-17 upon DDX3X stimulation. Surprisingly, the CD133+ melanoma cell-specific CD4+ T cells produced even more cytokines upon DDX3X stimulation than upon CD133+ melanoma cell-antigen stimulation. These results suggest that DDX3X possesses immunogenic MHC class II-restricted epitopes.
Vaccination with DDX3X conveyed protective immunity against parental melanoma cells
To examine whether protective immunity against B16 melanoma could be induced by vaccination with synthesized DDX3X proteins, DC pulsed with DDX3X or ovalbumin (OVA) at 5 μg/mL, or co-cultured with irradiated CD133+ tumor cells for 8 h, were subcutaneously (s.c.) injected in the right flank of the mice. Fourteen days later, the mice were s.c. inoculated in the midline of the abdomen with 2 × 106 parental melanoma cells. As shown in Fig. 3a, b, tumor growth was significantly more suppressed in the mice that received the DDX3X-pulsed DC vaccination compared to the mice that received either no treatment or treatment with OVA-pulsed DCs. Furthermore, the mice vaccinated with DCs pulsed with DDX3X exhibited significantly more potent protective immunity than did mice injected with DCs co-cultured with irradiated CD133+ tumor cells.

a, b Vaccination with synthesized DDX3X protein with DCs induced protective immunity against parental melanoma cells. Challenge of mice with parental B16 cells after immunization with DCs pulsed with DDX3X, ovalbumin (OVA) at 5 μg/mL, 5,000 cGy-irradiated parental, or CD133+ B16 melanoma cells for 8 h. DCs were isolated as CD11c+ cells with CD11c microbeads and autoMACS™ after 8-h culture. One million CD11c+ DCs were subcutaneously administered to B6 mice. Two weeks after immunization, the mice were subcutaneously inoculated along the midline of the abdomen with 2 × 106 parental B16 cells. Each group contained 5 mice. *P <0.05 and **P < 0.01. c, d, e Vaccination with synthesized DDX3X protein with DCs induced therapeutic antitumor reactivity against established skin melanoma. Parental B16 melanoma cells (2 × 105) were subcutaneously inoculated along the midline of the abdomen. On days 2, 9, 16 following tumor inoculation, 1 × 106 DCs pulsed with DDX3X or OVA at 5 μg/mL were subcutaneously injected at the mice’s right flanks. a Each group contained 5 mice. d Each group contained 12 mice. The tumor growth curve of each mouse is depicted in Fig. 4e. *P < 0.05 and **P < 0.01
Vaccination with DDX3X exhibited therapeutic efficacy against established skin tumors
We further tested if vaccination with DDX3X had therapeutic efficacy against established tumors. On days 2, 9, and 16 after s.c. tumor inoculation of 1 × 106 parental melanoma cells in the midline, 1 × 106 DCs were s.c. injected in the right flank. In the DDX3X-pulsed, DC-vaccinated mice, skin tumor growth was significantly suppressed (Fig. 3c, d). As shown in Fig. 3e, we found that 6 of 12 mice vaccinated with DDX3X-pulsed DC were eventually cured. All the mice that received no treatment or were vaccinated with OVA-pulsed DC died of the tumor.
Vaccination with CD133+ CSC that lacked DDX3X failed to induce antitumor immunity
It has been shown previously that B16 melanoma cells possess a number of immunogenic proteins. To elucidate the significance of DDX3X for the immunogenicity of CD133+ melanoma, we established CD133+ melanoma cells lacking DDX3X (Fig. 4a). As shown in Fig. 4b, mice vaccinated with CD133+ parental cells or CD133+ mock-transfectant tumor cells exhibited effective protective immunity. In contrast, vaccination with CD133+ tumor cells lacking DDX3X failed to induce antitumor protective immunity.

a Immunoblotting analyses of DDX3X expression in DDX3X-knockdown CD133+ B16 cells, mock-shRNA CD133+ B16 cells, and CD133+ B16 cells. b Protective antitumor immunity induced by DDX3X-knockdown CD133+ B16 tumor cell vaccination was significantly inferior to that induced by mock-shRNA CD133+ B16 or CD133+ B16 tumor cell vaccination. A total of 5,000 cGy-irradiated mock-shRNA and DDX3X knockdown CD133+ B16 cells were co-cultured with DCs for 8 h. One million CD11c+ cells purified with CD11c microbeads and autoMACS™ were subcutaneously administered to B6 mice. Two weeks after immunization, mice were subcutaneously inoculated along the midline of the abdomen with 2 × 106 parental B16 cells. Each group contained 5 mice. *P <0.05 and **P <0.01. c Efficacy of the antitumor therapeutic effect mediated by DDX3X-draining LN T cells. DCs were pulsed with synthesized DDX3X protein at 5 μg/mL for 8 h and isolated as CD11c+ cells. CD62Llow T cells were isolated as antigen-primed T cells from LNs draining DDX3X/DC or DCs. LN T cells were cultured for 5 days as described in the “Materials and methods” and intravenously infused into the mice bearing 2-day-established B16 subcutaneous tumors after sublethal whole body irradiation (500 cGy). Ten million T cells were infused intravenously. Each group contained 5 mice. *P < 0.05 and **P < 0.01. d Efficacy of the antitumor therapeutic effect mediated by DDX3X-draining CD4+ or CD8+ LN T cells. CD62Llow LN T cells were isolated as antigen-primed T cells and were further purified as CD4+ or CD8+ T cells with magnetic beads, following the manufacturer’s instructions. After the LN T cells were purified, they were cultured for 5 days, as described in the “Materials and methods”, and intravenously infused into the mice bearing 2-day-established B16 subcutaneous tumors after sublethal whole body irradiation (500 cGy). Ten million T cells were infused intravenously. Each group contained 5 mice. *P < 0.05, and **P < 0.01
DDX3X/DC-primed CD4+ T cells from draining LNs mediate potent antitumor therapeutic efficacy
To elucidate that T cells mediated the antitumor reactivity that was induced by DDX3X vaccination, CD62Llow T cells isolated from draining LNs were infused intravenously (i.v.) into mice with 2-day, established skin melanoma tumors. This was done following sublethal, whole body irradiation (500 cGy). We found that T cells primed with DDX3X exhibited antitumor reactivity, resulting in significant suppression of skin tumor growth (Fig. 4c). We then examined whether CD4+ or CD8+ T cells preferentially mediated antitumor reactivity. Ten million isolated CD8+ LN T cells were injected, but showed no significant antitumor reactivity. In contrast, CD4+ DDX3X-specific T cells were found to mediate potent antitumor therapeutic efficacy, thus curing the melanoma (Fig. 4d).
DDX3X is expressed in human cancer cells
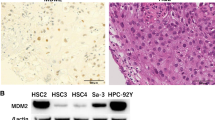
We next investigated whether DDX3X is expressed in human cancer cells, using the following cell lines: 87.5 and S2 (small cell lung cancer), HCT116 (colon cancer), A549 (non-small cell lung cancer), WM115 (melanoma), and MCF7 (breast cancer). The 87.5 cells and HCT116 expressed CD133 (Fig. 5). The 87.5 cells proliferated as floating aggregates and easily formed tumor spheres. MCF7 cells contained the CD44+ and CD24−/low population. Immunoblotting analyses revealed that all of the examined cancer cells expressed DDX3X and that 87.5, HCT116, and MCF7 cells strongly expressed DDX3X (Fig. 5). In contrast, normal human epidermal keratinocytes (NHEK), normal human mammary epithelial cells (HMEC), and normal human bronchial epithelial cells (NHBE) only faintly expressed DDX3X (data not shown). These results suggest that, in addition to its expression in murine melanoma stem cells, DDX3X is expressed in various human cancer cells.

Detection of DDX3X in human cancer cells. a Whole cell lysates were extracted from 87.5 and S2 (human small cell lung cancer lines), HCT116 (a human colon cancer), A549 (a human, non-small cell lung cancer), WM115 (a human melanoma), and MCF7 (a human breast cancer) cells. Immunoblots from tumor cells were probed with antibodies against DDX3X and β-actin. b Human cancer cell lines, that is, 87.5 and S2 (small cell lung cancer lines), HCT116 (colon cancer cell line), A549 (non-small cell lung cancer cell line), WM115 (melanoma cell line), and MCF7 (breast cancer cell line) were examined for the expression of putative CSC markers. It was found that 87.5 and HCT116 cells expressed CD133, whereas the other cancer cells did not. MCF7 cells contained CD44+ and CD24−/low populations
Discussion
Our proteome analyses detected 4 proteins that are preferentially expressed in CD133+ B16 melanoma, which is characterized by features of cancer stem cells, including high tumorigenicity, non-adherent growth in tumor spheres, and the ability to differentiate into either CD133+ or CD133− tumor cells. A Mascot search based on mass spectrometry (MS/MS) protein data identified one of the 4 proteins as DDX3X, a member of the DEAD-box family of proteins named after a conserved D-E-A-D sequence that displays RNA-dependent ATPase and ATP-dependent RNA helicase activities. The DDX3X gene is located on the X chromosome and is evolutionarily well conserved from yeast to humans [13]. Although DDX3X was reported to have tumor-suppressor functions in virus-associated cancer cells [20, 21], it has been revealed that DDX3X plays an oncogenic role by facilitating β-catenin signal transduction and induces CSC-like features, such as an epithelial-mesenchymal transition accompanied by loss of E-cadherin, increased motility, and invasive properties with up-regulated snail expression in breast cancer line cells [19, 22, 23]. Moreover, recent whole-exome analyses identified that DDX3X is one of the genes that drive medulloblastoma, which is similar in appearance and differentiation potential to neural stem and progenitor cells [14, 24]. DDX3X knock down resulted in loss of neurosphere formation ability. Consistent with these reports, we found that CD133+ melanoma cells were no longer characterized by high motility, the ability to grow in tumor spheres, or colony formation in soft-agar assay following knock down of DDX3X (data not shown). Thus, it is likely that DDX3X plays an essential role in stem cell functions in cancer cells.
Our study further revealed that DDX3X is an immunogenic protein. That is, it possesses highly immunogenic MHC class II-restricted epitopes. This was shown when CD4+ T cells primed by vaccination with irradiated CD133+ melanoma cells were found to secrete even more cytokines upon DDX3X stimulation as compared to CD133+ melanoma cell-antigen stimulation alone. In other words, it is likely that the majority of T cells primed by CD133+ melanoma cell vaccination recognized DDX3X. Consistent with this observation, CD133+ melanoma cells with DDX3X knock down lost immunogenicity and were thus unable to mount antitumor protective immunity.
DDX3Y, a homologue of DDX3X with 92 % homology located on the Y chromosome, is a male-specific minor histocompatibility antigen. Male leukemia patients who received HLA-identical hematopoietic stem cell transplantation from female donors reportedly showed DDX3Y-specific T cell responses and antibody production [25–29]. Since DDX3-specific immune responses were detected in approximately 50 % of these leukemia patients, it is likely that DDX3 possesses immunogenic MHC class I- and II-restricted epitopes. Furthermore, it was indicated that DDX3-specific T cells mediated graft versus leukemia effects and that DDX3-specific T cells eradicated leukemia stem cells [25]. Interestingly, leukemia patients primed by T cell clones responded similarly to both corresponding epitopes of DDX3Y and DDX3X. Thus, immunogenic epitopes, which can induce graft versus leukemia effects, were shared between DDX3X and DDX3. However, disparities between DDX3Y and DDX3X triggered female donor-T cell priming in male leukemia patients. Our study revealed that human small cell lung cancer, colon cancer, and breast cancer cells with CSC markers expressed a high level of DDX3X (Fig. 5). Furthermore, we detected DDX3X-specific T cells in approximately 50 % of patients with limited disease, small cell lung cancer, but not in extended disease patients or healthy donors at all (unpublished data).
We also found that CD4+ T cells primed by DDX3X vaccination discriminated CD133+ from CD133− melanoma cells and mediated potent antitumor immunity. On the other hand, CD8+ T cells primed with DDX3X vaccination responded to both CD133+ and CD133− tumor cells. According to proteome analyses, CD133− melanoma cells expressed half the amount of DDX3X as did CD133+ melanoma cells. It is possible that CD4+ but not CD8+ T cells could distinguish CD133+ melanoma cells since CD4+ T cells require more TCR engagement for activation than that required by CD8+ T cells.
In summary, this study represents the first demonstration of DDX3X as an immunogenic antigen capable of mounting antitumor immune responses. Our results show that DDX3X could be used as a therapeutic target antigen for antitumor immunotherapy, which may be very useful in clinical settings.
References
Li Y, Laterra J (2012) Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res 72:576–580. doi:10.1158/0008-5472.CAN-11-3070
Sharma SV, Lee DY, Li B et al (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141:69–80. doi:10.1016/j.cell.2010.02.027
Rappa G, Fodstad O, Lorico A (2008) The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells 26:3008–3017. doi:10.1634/stemcells.2008-0601
Milas L, Raju U, Liao Z, Ajani J (2005) Targeting molecular determinants of tumor chemo-radioresistance. Semin Oncol 32:S78–S81. doi:10.1053/j.seminoncol.2005.04.028
Diehn M, Clarke MF (2006) Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst 98:1755–1757. doi:10.1093/jnci/djj505
Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH (2005) ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 65:4320–4333. doi:10.1158/0008-5472.CAN-04-3327
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. doi:10.1038/nature05236
Malanchi I, Peinado H, Kassen D et al (2008) Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 452:650–653
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R (2007) Identification and expansion of human colon-cancer-initiating cells. Nature 445:111–115. doi:10.1038/nature05384
Kagamu H, Shu S (1998) Purification of L-selectin(low) cells promotes the generation of highly potent CD4 antitumor effector T lymphocytes. J Immunol 160:3444–3452
Fujita N, Kagamu H, Yoshizawa H et al (2001) CD40 ligand promotes priming of fully potent antitumor CD4(+) T cells in draining lymph nodes in the presence of apoptotic tumor cells. J Immunol 167:5678–5688
Miyabayashi T, Kagamu H, Koshio J et al. (2011) Vaccination with CD133(+) melanoma induces specific Th17 and Th1 cell-mediated antitumor reactivity against parental tumor. Cancer Immunol Immunother. doi:10.1007/s00262-011-1063-x
Lahn BT, Page DC (1997) Functional coherence of the human Y chromosome. Science 278:675–680
Pugh TJ, Weeraratne SD, Archer TC et al (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488:106–110. doi:10.1038/nature11329
Stransky N, Egloff AM, Tward AD et al (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333:1157–1160. doi:10.1126/science.1208130
Wang L, Lawrence MS, Wan Y et al (2011) SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365:2497–2506. doi:10.1056/NEJMoa1109016
Hiura T, Kagamu H, Miura S, Ishida A, Tanaka H, Tanaka J, Gejyo F, Yoshizawa H (2005) Both regulatory T cells and antitumor effector T cells are primed in the same draining lymph nodes during tumor progression. J Immunol 175:5058–5066
Watanabe S, Kagamu H, Yoshizawa H, Fujita N, Tanaka H, Tanaka J, Gejyo F (2003) The duration of signaling through CD40 directs biological ability of dendritic cells to induce antitumor immunity. J Immunol 171:5828–5836
Botlagunta M, Vesuna F, Mironchik Y et al (2008) Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene 27:3912–3922. doi:10.1038/onc.2008.33
Wu DW, Liu WS, Wang J, Chen CY, Cheng YW, Lee H (2011) Reduced p21(WAF1/CIP1) via alteration of p53-DDX3 pathway is associated with poor relapse-free survival in early-stage human papillomavirus-associated lung cancer. Clin Cancer Res 17:1895–1905. doi:10.1158/1078-0432.CCR-10-2316
Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH (2006) DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res 66:6579–6588. doi:10.1158/0008-5472.CAN-05-2415
Mani SA, Guo W, Liao MJ et al (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715. doi:10.1016/j.cell.2008.03.027
Sun M, Song L, Zhou T, Gillespie GY, Jope RS (2011) The Role of DDX3 in regulating snail. Biochim Biophys Acta. doi:10.1016/j.bbamcr.2011.01.003
Fan X, Eberhart CG (2008) Medulloblastoma stem cells. J Clin Oncol 26:2821–2827. doi:10.1200/JCO.2007.15.2264
Rosinski KV, Fujii N, Mito JK et al (2008) DDX3Y encodes a class I MHC-restricted H-Y antigen that is expressed in leukemic stem cells. Blood 111:4817–4826. doi:10.1182/blood-2007-06-096313
Porcheray F, Miklos DB, Floyd BH et al (2011) Combined CD4 T-cell and antibody response to human minor histocompatibility antigen DBY after allogeneic stem-cell transplantation. Transplantation 92:359–365. doi:10.1097/TP.0b013e3182244cc3
Zorn E, Miklos DB, Floyd BH, Mattes-Ritz A, Guo L, Soiffer RJ, Antin JH, Ritz J (2004) Minor histocompatibility antigen DBY elicits a coordinated B and T cell response after allogeneic stem cell transplantation. J Exp Med 199:1133–1142. doi:10.1084/jem.20031560
Miklos DB, Kim HT, Zorn E et al (2004) Antibody response to DBY minor histocompatibility antigen is induced after allogeneic stem cell transplantation and in healthy female donors. Blood 103:353–359
Vogt MH, van den Muijsenberg JW, Goulmy E et al (2002) The DBY gene codes for an HLA-DQ5-restricted human male-specific minor histocompatibility antigen involved in graft-versus-host disease. Blood 99:3027–3032
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan. Hiroshi Kagamu received research fund from Otsuka Pharmaceutical Co. Ltd. (Tokyo, Japan).
Conflict of interest
All other authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jun Koshio and Hiroshi Kagamu have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Koshio, J., Kagamu, H., Nozaki, K. et al. DEAD/H (Asp–Glu–Ala–Asp/His) box polypeptide 3, X-linked is an immunogenic target of cancer stem cells. Cancer Immunol Immunother 62, 1619–1628 (2013). https://doi.org/10.1007/s00262-013-1467-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-013-1467-x