
Abstract
There is ample evidence that the presence of tumor-infiltrating T lymphocytes is associated with a favorable prognostic in patients. These observations suggest that a limiting step to immune resistance and immunotherapy could be the capacity of tumor-specific T cells to reach tumor bed. In this article, we review some factors that may influence this infiltration, and in particular the nature of the vasculature, the expression of chemokines or tumor antigens and the presence of dendritic cells and CD4+ T lymphocytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
There is an increasing evidence that tumor infiltration by T lymphocytes is associated with a good prognosis, although the clinical outcome may depend on the composition and architecture of the immune infiltrate.
Large clinical studies have underscored the prognostic and predictive impact of the immune infiltrates for colorectal, ovarian, breast cancers, and melanomas. In human colorectal cancer, the presence of CD8+ T cells, expressing granzyme+ cytoplasmic granules and highly proliferating, within cancer cell nests was associated with a better survival of patients [1]. Galon et al. reported that human colorectal cancers with a high density of memory T cells were associated with the absence of pathological evidence of early metastatic invasion and with increased survival [2]. In a subsequent study, the authors measured the levels of mRNA for inflammatory and immunosuppressive molecules in colorectal tumors and found increased levels of CD8α, granzyme, granulysin, T-bet, IFN-γ, and IRF-1 in tumors from patients who had not relapsed [3]. Of note, tumors from patients without recurrence had higher immune cell densities in the center and the invasive margin at stages II and III, suggesting a beneficial effect throughout tumor progression. A recent study confirmed the role of tumor-infiltrating CD45RO+ whose density was associated with longer survival in patients with colorectal cancer [4]. Similarly, the presence of intratumoral T cells was shown to correlate with favorable clinical outcome in advanced ovarian carcinoma: The 5-year overall survival was 38 % among patients whose ovarian tumors contained T cells and 4.5 % among patients whose tumors contained no T cells [5]. Sato et al. [6] further reported that the presence of intraepithelial CD8+ TIL and high CD8+/CD4+, CD8+/regulatory T cell ratios were associated with improved survival in ovarian cancer. The biological significance of lymphocytic infiltration in malignant melanomas is still unclear, but several observations suggest a correlation between regression and amplification of T cells in situ [7, 8]. Ferradini et al. [9] have characterized the molecular structure of the T cell receptor β chain of TILs in a case of regressive melanoma and reported that 84 % of the Vβ16 transcripts analyzed in the tumor corresponded to the same cDNA clone, but were different in PBL from the same patient. Concerning the Vβ4 transcripts, one cDNA clone represented about 45 % of the transcripts in the tumor, while all transcripts in PBL were different. In resected advanced-stage melanoma patients, treated by autologous tumor cell vaccinations, TAA-specific TIL could be detected in 75 % of tumor samples. Of note, the median survival was 22.5 and 4.5 months in case of the presence or absence of detectable TAA-specific TIL, respectively.
Interestingly, there is evidence that the infiltration of immune cells is not only a good prognostic factor (as being involved in immunosurveillance) but also could be predictive of the efficacy of conventional chemotherapies in patients. Denkert et al. [10] identified a subgroup of breast carcinomas characterized by tumor-associated lymphocytes and a particularly strong response to chemotherapy. Other studies have linked immune-related genes to response to chemotherapy [11, 12]. Several mechanisms may be involved in the effect of conventional chemotherapy on immune resistance and include reduction of the tumor mass, lymphopenia-driven homeostatic activation, enhanced antigen presentation, reversal of tumor-induced immune tolerance, etc. [13]. Interestingly, Zitvogel et al. have shown that chemotherapy can cause immunogenic cancer cell stress or death, thereby creating a cancer vaccination effect. In particular, depending on the cell death inducer, tumor cells can express factors that favor their uptake by dendritic cells (as the chaperone calreticulin which translocates from the lumen of the endoplasmic reticulum to the plasma membrane), or antigen presentation by dendritic cells (as the high mobility group box 1 protein, or HMGB1, a chromatin-binding protein that is released by dying cells in some conditions and interacts with TLR4 to favor the trafficking of antigen to the antigen-presenting compartment) [13, 14]. It should be noted that the contribution of immunity to chemotherapy may be one mechanism among others and requires specific factors: defined cytotoxic agents, tumor properties, and host immune characteristics.
A recent report raises some doubt on the hypothesis that the adaptive immune response dictates the efficacy of chemotherapy [15]. Indeed, the authors found that the adaptive immune system was dispensable for the therapeutic efficacy of three chemotherapeutics in two spontaneous mammary mouse tumors. They conclude that the immune system may contribute to chemotherapy response only in transplanted tumors which are more immunogenic. Nevertheless, the observations in patients with cancer suggest that chemotherapy may, in some conditions, reactivate pre-therapeutic immunosurveillance mechanism.
Numerous studies in the mouse have indeed illustrated the capacity of the immune system to maintain cancer cells in equilibrium for extended period of time and have highlighted the critical function of IFN-γ and cytotoxic responses to control tumor growth [16]. Accordingly, in patients with colorectal cancer, unsupervised hierarchical clustering revealed functional clusters of genes associated with T helper subsets. Patients with elevated expression of the Th17 cluster had a poor prognosis, whereas high Th1 and cytotoxic gene levels were associated with a good outcome [17].
Collectively, these observations strongly suggest that immunosurveillance in patients correlates with tumor infiltration and in particular with the trafficking of CD8+ T lymphocytes to the tumor bed. The factors that influence the movement of immune effector cells inside the tumor are still poorly defined. In this article, we will review some mechanisms governing this migration that have been explored mainly in murine tumor models.
Tumor vasculature
A critical step in the movement of lymphocytes from lymphoid organs into peripheral tissues, including tumors, is the adhesion of lymphocyte to vascular endothelium. A recent report examined the presence of high endothelial venules (HEV, specialized post-capillary venules found in lymphoid organs that allow high levels of lymphocyte extravasation from the blood) in various human solid tumors [18]. The authors show that human tumors (melanomas, breast, ovarian, colon, and lung carcinomas) contained blood vessels resembling HEV, which appear specifically located into lymphocyte-rich tumor areas. A high density of HEVs was associated with tumor-infiltrating T and B cells in breast tumors and correlated with higher expression of genes involved in lymphocyte migration, Th1 adaptive immune response and cytotoxic function. These features were associated with a favorable clinical outcome. Another report demonstrated the presence of tertiary lymphoid structures in the tumor stroma in human lung cancers, which correlated with long-term survival in patients [19]. Of note, PNAd+ HEV were detected in these tumors, exclusively associated with the tertiary structures [20]. These HEV were shown to express adhesion molecules known to interact with the integrins alpha chains related to T cell infiltration (see later discussion in “Adhesion molecules and chemoattractants” section).
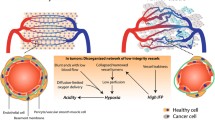
However, there is clear evidence that tumor vessels can become abnormal in their structure and function [21] (Fig. 1). A role for VEGF as a negative regulator of vessel maturation has been reported: VEGF appears to play opposing roles in angiogenesis, stimulating directly endothelial cell proliferation and migration, and inhibiting pericyte coverage of nascent vascular sprouts, leading to vessel destabilization [22]. The authors examined the vasculature of murine fibrosarcomas expressing or not VEGF and noticed that VEGF−/− tumors produced mature vessels with pericyte coverage, whereas WT tumors developed an immature vasculature. To test the role of myeloid-cell-derived VEGF in tumor progression in mice (tumors express VEGF-A released by both malignant and stromal cells), Stockmann et al. [23] created an in vivo, cell-lineage-specific, targeted deletion of Vegfa and reported that the vasculature in solid tumors lacking myeloid-derived VEGF-A was normal and resulted in accelerated tumor progression. Thus, myeloid-derived VEGF-A causes tumorigenic alteration of vasculature. However, the clinical effect of antiangiogenic therapy is more challenging than anticipated. Indeed, vessel normalization may increase tumor responses to conventional therapies and to immunotherapy, but antiangiogenic therapy may also increase hypoxia and create a pro-tumorigenic inflammatory state, thereby aggravating tumor metastasis. Of note, Shrimali et al. [24] demonstrated that disrupting VEGF/VEGFR-2 signaling could increase extravasation of adoptively transferred T cells into established B16 melanoma and enhance their antitumor efficacy.

Schematic representation of tumor infiltration. Tumor clinical outcome is often associated with the presence of tumor-infiltrating lymphocytes (TILs), tertiary lymphoid structures and/or high endothelial venules (HEVs). However, during tumor progression, tumor vessels may become heterogenous and tortuous, which could result from (1) production by tumor and/or myeloid cells (MC) of VEGF which induces abnormal differentiation and proliferation of endothelial cells and is a negative regulator of pericyte function [21, 22]; (2) secretion of endothelin-1 (ET-1) by tumor cells, which upon interaction with its ligand (ETBR) expressed by tumor abnormal vessels, inhibits T cell adhesion to endothelium by NO production and downregulation of ICAM-1 molecules [24]. EC endothelial cells, VEGF vascular endothelial growth factor, VEGFR-P VEGF receptor phosphorylated
An elegant study by Buckanovich et al. [25] identified the endothelin B receptor as responsible for the endothelial barrier to T cell homing to tumors. Using transcriptional profiling of microdissected tumor endothelial cells from human ovarian cancers, they showed that specific endothelial molecules were associated with the presence or absence of lymphocyte infiltration. In particular, the endothelin B receptor was highly expressed in 80 % of tumors lacking intraepithelial TILs. The 5-year survival rate was 6.6 % for patients bearing tumors expressing high endothelin B receptor mRNA levels and 52.2 % for individuals expressing lower levels. The authors further show that endothelin B receptor blockade increased T cell adhesion to human endothelium in vitro through upregulation and relocalization of ICAM-1. Finally, they showed that receptor blockade in mice in vivo led to an increased influx of TILs and a higher expansion of tumor-specific T cells in ID8 ovarian tumors. Accordingly, other reports have demonstrated that isolated tumor endothelial cells had suppressed levels of ICAM-1 as compared from normal mouse tissues [26, 27].
In the mouse, two additional genes have been shown to affect vascular architecture. Using a mouse model of pancreatic islet carcinogenesis, Hamzah et al. [28] highlighted the role of the regulator of G-protein signaling 5 (Rgs5), a gene expressed by pericytes in numerous tumors, as a master gene responsible for the abnormal tumor vasculature. Rgs5-deficient mice displayed normalized vasculature and were highly responsive to therapeutic vaccination by T cells. Finally, the thrombospondin-1 (TSP-1) has been shown to inhibit angiogenesis through induction of endothelial apoptosis in vitro and in vivo in lung metastases of murine melanoma [29]. TSP-1 is upregulated in the platelets of tumor-bearing mice, which may constitute an early critical host response to suppress tumor growth. TSP-1, produced by CD4+ T cells, has been shown to favor the shutdown of angiogenesis and cellular senescence upon inactivation of MYC oncogene in a mouse model of T cell lymphoma [30].
Adhesion molecules and chemoattractants
Recruitment of blood cells to lymph nodes requires a specific set of adhesion molecules and chemoattractants. Among others, the L-selectin/CD62L mediates rolling on HEV, the integrins α4β7 and LFA-1 mediate a firm adhesion and junctional adhesion molecules, such as CD31 (PECAM), CD99, and JAM1, favor their extravasation. The chemokines CCL19, CCL21 direct the movement of lymphocytes expressing CCR7. These molecules guide naive and central memory lymphocytes that constantly circulate between blood and peripheral lymphoid organs [31]. The role of adhesion molecules has been addressed in transgenic mice expressing an oncoprotein in islet β cells, leading to β cell hyperplasia with subsequent progression to tumors and lymphocytic infiltration [32]. Endothelial ligands for L-selectin and α4β7 were upregulated only in infiltrated islets and correlated with tumor immunity. In inflammatory conditions, lymphocytes activated in the lymphoid organs migrate to the pathological sites (inflammatory foci, infected tissues, and tumor, …) using different receptor/ligands pairs: effector lymphocytes express P-selectin glycoprotein ligand 1 (PSGL-1) that binds to P- and E-selectins present on the surface of inflamed endothelium, and chemokine receptors CXCR3 and/or CCR5 which bind to chemokines CXCL9/10/11 or RANTES (also named CCL5), respectively.
It is generally believed that tumor-specific T lymphocytes activated in the lymph nodes reach the tumor through inflammation-induced ligand/receptor pairs. It is likely that tumor-specific T lymphocytes are activated in the lymph nodes by CD8α+ dendritic cells specialized in cross-presentation, as elegantly demonstrated in Batf3−/− mice (which lack the CD8α+ DC subset and are not able to reject immunogenic tumor) [33]. However, naïve T lymphocytes are able to infiltrate tumors in some conditions and to undergo activation at the tumor site. Schrama et al. [34] showed that targeting of lymphotoxin-α to mouse melanoma elicited the formation of a lymphoid-like tissue comprising high endothelial venules, a potential entry for naïve T cell infiltrates (one-third of the blood vessels in the tumors had HEV morphology, i.e., endothelial cells of cuboidal shape). Priming occurred in the tumor, as assessed by the presence of clonally expanded T cells within the tumor, which were not detected in the lymph nodes. Yu et al. [35] reported that forced expression of LIGHT in Ag104 murine fibrosarcoma induced a massive infiltration of naïve T lymphocytes. LIGHT is a tumor-necrosis factor superfamily member and may overcome the tumor barrier by attracting and activating T lymphocytes into the tumor through LTβR (lymphotoxin-β receptor) and HVEM (herpes simplex virus glycoprotein D), respectively, leading to tumor rejection. Interestingly, recent evidence suggests that naïve T lymphocytes can also reach unmanipulated tumors [36]. The recruitment of naïve T cells occurred in various murine tumor types and for T cell with different avidities in the absence of any intentional inflammation and resulted in their proliferation and differentiation into cytotoxic effectors. An interesting question is whether the movement of naïve T cells was stochastic, or directed toward tertiary lymphoid structures present in the tumor mass.
Therefore, the same sets of molecules described previously (see Table 1) may direct the migration of naïve and effector T cells to lymphoid structures and inflammatory sites in the tumor, respectively. Of note, several reports support a critical role for CXCR3, a receptor for CXCL9/10/11 chemokines expressed by still undefined cells within tumors. However, specific features have been described, which may impede effector lymphocyte infiltration in tumors: (1) the downregulation of ICAM-1 on tumor vessels (see previous discussion in “Tumor vasculature” section), (2) the recruitment of regulatory T cells through CCL22 production by tumor cells (human breast carcinoma) in the presence of innate effector cells [37], (3) the nitrosylation of the chemokine receptor CCL2 in human and mouse tumors which can prevent T lymphocyte, but not MDSC, migration thereby enhancing the production of the radical NO2 inside the tumor [38]. Nitrosylated CCL2 was found in human prostate and colon carcinomas as well as in several murine tumors and blocking the production of peroxynitrite reversed the block in T cell trafficking, leading to enhanced tumor rejection in three mouse models (colon carcinoma C26GM, EG7-OVA, a spontaneous prostate cancer).
A recent report demonstrates that chemotherapy may induce intratumoral expression of T cell-attracting chemokines in cancer cells [39]. The authors used the RETAAD mice which develop spontaneous uveal melanomas that metastasize to the skin and visceral organs. A strong antitumor response is developed which controlled visceral metastases but had no effect on cutaneous tumors. The authors showed that the median percentage of CD3+ T cells was 2.7 times higher in visceral metastases than in spontaneous cutaneous tumors, but that T cells highly infiltrated transplanted cutaneous tumors. The comparison of the repertoire of chemokines expressed in RETAAD versus transplanted B16 tumors revealed a differential expression of chemokines, with a 105- and 42-fold greater expression of Cxcl9 and Cxcl10 in B16 tumors, respectively. Of note, three chemotherapeutic drugs induced chemokine (CXCR3 ligands and ccl5) expression and T cell infiltration in Rag−/− mice bearing subcutaneous Melan-ret tumors as well as in patients with melanoma. This enhanced expression translates into improved tumor control and prolonged survival.
Tumor antigens
The dynamics of tumor-infiltrating T lymphocytes with tumor cells has been analyzed in tumors using two-photon microscopy. Two reports demonstrate that recognition of cognate antigen within tumors is critical for optimal T cell infiltration and target cell interactions. Mrass et al. [40] reported that TIL migrated randomly both in the EL-4 explants and in tumors in situ but that recognition of cognate antigen was required for the maintenance of a highly active migratory phenotype. Boissonnas et al. [41] further showed that CTLs transiently stopped moving only in OVA-expressing EG7 tumors. Of note, deep tumor infiltration during rejection required antigen expression, as OT-I cells infiltrating EL4 tumors (not expressing OVA) remained in the periphery.
Dendritic cells
An interesting question for cancer immunotherapy is how the in vivo tissue distribution of antigens recognized by CD8+ T lymphocytes may regulate the imprinting of their homing properties. Calzascia et al. [42] monitored the phenotypic changes of tumor-specific T cells from the priming phase to the effector phase in tumors. They implanted the MC57-GP fibrosarcoma (expressing the lymphocytic choriomeningitis virus glycoprotein) intracerebrally, subcutaneously or intraperitoneally and adoptively transferred CFSE-labeled naïve CD8+ T cells from P14 TCR transgenic mice recognizing the H-2Db-restricted GP epitope. Different sets of lymphoid tissues exhibited proliferation of CFSE-labeled cells and three different adhesion patterns characterized GP-specific CD8+ T cells, which proliferated in cervical versus inguinal versus mesenteric LN. The site of tumor implantation rather than the nature of the LN appeared to determine the homing phenotype, as subcutaneous implantation of the tumor imprinted a similar phenotype on P14 CD8+ T cells dividing in cervical as that induced in inguinal LN after subcutaneous implantation in the flank. The authors propose a model of phenotypic imprinting that is dictated not in the LN but rather upstream at the site of antigen capture by cross-presenting APCs. This conclusion is in agreement with previous reports that underline the role of dendritic cells in the selective imprinting of tissue-homing T cells [43–45].
Although their role has not been illustrated in tumor models, dendritic cells may also regulate vascular growth. Thus, Webster et al. [46] have reported that immunization with ovalbumin in CFA induced endothelial cell proliferation in draining lymph nodes. This early event, which occurred 24 h after immunization, was strongly diminished when dendritic cells were depleted by diphtheria toxin injection (in CD11c-DTR mice). Their observations suggest that DCs which migrate to the lymph nodes after activation in the periphery, activate endothelial cell proliferation by inducing the entry through HEVs of circulating cells that directly or indirectly increase LN VEGF levels.
CD4+ T cells
It has been known for decades that CD4+ T “helper” cells are critical for the priming of CD8+ T lymphocytes and their development into effector and memory cells. Interestingly, CD4+ T cells seem to act also as gate-keepers of CTL entry into infected tissues. Iwasaki and colleagues reported that CD4+ T cells controlled the migration of CTL by altering the microenvironment of the HSV-2 infected tissue in mice [47]. When inoculated into the vaginal cavity, HSV-2 replicates predominantly in the mucosal epithelial cells and the viral infection remains localized, rendering this infectious model suitable to study lymphocyte infiltration. Both CD4+ and CD8+ T cells were primed in the draining lymph nodes and CD8+ T lymphocytes migrated to the infection site, in the presence but not the absence of CD4+ T cells. The authors further show that CTL recruitment was dependent on (1) CXCR3 expression by CD8+ T cells and (2) IFN-γ production by CD4+ T cells leading to the production of CXCL9 and CXCL10 in the infected tissue. Another report demonstrates that activated CD4+ T cells cause CD8+ T cell accumulation depending on inflammation-induced CCR5 expression on CD8+ T cells and production of CCL3 and CCL4 by dendritic cells and/or CD4+ T cells [48]. Of note, this chemokine-guided migration will favor the accumulation of CD8+ T lymphocytes in sites of productive CD4+ T cell–dendritic cell interaction. In addition, CD4+ T cells may help primary CD8+ T cell responses by supporting expansion of the arteriole feeding the reactive lymph node, resulting in increased cellular input. Whether the arteriole remodeling is mediated by DC and/or CD4+ T cells is unknown at present [49].
A few reports confirm a role for CD4+ T cells in the tumor microenvironment (Fig. 2). Wong et al. [50] showed that the presence of CD4+ T cells facilitated accumulation of CD8+ T cells, including low-avidity tumor-specific cells, within spontaneous pancreatic tumors in mice. Bos and Sherman [51] examined the intratumoral accumulation of tumor-specific CD8+ T cells in a mouse model of pancreatic neuroendocrine tumor. The presence of CD4+ T cells induced a strong inflammatory environment and correlated with enhanced infiltration of CD8+ T cells. Several IFN-γ-induced chemokines seemed to be involved: CXCL9, 10, CCL2, 3 and 5.

Potential roles of CD4+ T lymphocytes in chemokine production involved in the recruitment of tumor-specific CD8+ T lymphocytes. In several tumors in mice and humans, Th1 cells have been shown to produce IFN-γ which induces the expression of CXCL9 and CXCL10 in the tumor microenvironment. Similarly, in one pancreatic tumor model, CCL3 and CCL5 are induced by CD4+ T cells producing IFN-γ [48]. The role of Th17 cells is less clear: they may act as inducers of the expression of CCL2 and CCL20 in the tumor environment to recruit CD8+ T lymphocytes [51] and/or as activators of Th1 cells to produce IFN-γ and CXCL9/CXCL10 chemokines required for CD8+ T cell migration [52]
There is some evidence that Th17 cells may similarly promote effector T cell and NK cell trafficking to the tumor microenvironment [52], an observation consistent with the role described for IL-17 in mice vaccinated to M. tuberculosis [53]. Indeed, in wild-type but not IL-12p19-deficient mice, vaccination induced expression of the genes coding for CXCL9, 10, 11 (CXCR3 ligands). Treatment with anti-IL-17 neutralizing antibody inhibited the induction of the genes encoding all three chemokines and reduced the frequency of antigen-specific IFN-γ producing T cells in the lungs of vaccinated wild-type mice. In a mouse tumor model, IL-17A-deficient mice were shown to be more susceptible to developing lung melanoma and therapy using Th17 cells was remarkably efficient [54]. The authors further showed that Th17 cells induced lung cells to produce CCL2 and CCL20, which favor leukocyte homing to tumors. Of note, the predominant DCs recruited were CD8α+ DCs that are indispensable for cross-presentation. Kryczek et al. [55] studied the immune and clinical responses in 201 patients with ovarian cancer and showed that Th17 cells synergized with IFN-γ to stimulate CXCL9 and CXCL10 production in the tumor. A significant association was found between ascites IL-17 levels and survival. However, the role of Th17 in tumor resistance remains controversial and deserves further attention (see earlier discussion).
Conversely, impaired T cell homing and preferential recruitment of regulatory T cells have been described in many human cancers, leading to evasion to the immune response. Blood vessels of invasive squamous cell carcinomas of the skin do not express E-selectin and the tumors contained few CLA+ (ligand for E-selectin) T cells but a large population of Foxp3+ T cells. Of note, treatment with the TLR7/8 agonist imiquimod induced vascular E-selectin and infiltration of CLA+ T cells into the tumor [56]. A recent report suggests that the CD73, a surface ectonucleotidase expressed in many types of murine and human tumors, may control antitumor T cell homing to tumors [57]. Loss of CD73 in mice resulted in increased infiltration of CD8+ T cells and decreased Foxp3+ T cell infiltrates. Blockade of CD73 in vitro restored adhesion of T cells and blockade of CD73 (or inhibition of its enzymatic activity) increased antigen-specific T cell homing in vivo to EG7 and B16-SIY tumors, suggesting that CD73 may protect tumors from incoming specific effector cells.
Conclusion
Although cancer antigens have been discovered several decades ago in human melanomas, immunotherapy is still at an early stage. Several limiting steps may hamper the capacity of existing tumor-specific T lymphocytes to control tumor growth [58]. However, the correlation between tumor lymphocyte infiltration and favorable prognostic suggests that antigen expression and immunosuppressive mechanisms may not be the limiting factors. By contrast, the coordination of the events required to guide effector cells to the tumor bed is clearly more complex than expected and include the control of tumor vasculature, the expression of correct sets of adhesion molecules and chemoattractants (and their receptors) and the prevention of multiple inhibitory mechanisms [59]. An interesting question is whether active immunotherapy may boost tumor-specific T lymphocytes by creating a microenvironment which favors infiltration inside the tumor, as suggested by the increased proportion of antitumor (distinct from antivaccine) TIL after vaccination in melanoma patients [60]. Similarly, whether T cell infiltration is the cause or consequence of tumor regression cell in spontaneous or chemotherapy-induced regression is still a matter of speculation. Nonimmunological mechanisms may indeed result in decreased tumor progression and favor T cell migration in tumor bed. An in depth analysis of tumor biology (in particular the proliferation index) and the analysis of the T cell repertoire at different time points in the tumor and the draining lymph node, and their correlation with T cell infiltration may help clarify this issue.
References
Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H (1998) CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 58:3491–3494
Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc P-H, Trajanoski Z, Fridman W-H, Galon J (2005) Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 353:2654–2666
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc P-H, Trajanoski Z, Fridman W-H, Pagès F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (New York, N.Y.) 313:1960–1964
Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt J, Giovannucci E, Dranoff G, Fuchs CS, Ogino S (2010) Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol 222:350–366
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty P, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G (2003) Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348:203–213
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, Lele S, Chen Y, Ohtani H, Old LJ, Odunsi K (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Nat Acad Sci USA 102:18538–18543
Bogunovic D, O’Neill DW, Belitskaya-Levy I, Vacic V, Yu Y, Adams S, Darvishian F, Berman R, Shapiro R, Pavlick AC, Lonardi S, Zavadil J, Osman I, Bhardwaj N (2009) Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Nat Acad Sci USA 106:20429–20434
Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF (2009) Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69:3077–3085
Ferradini L, Mackensen A, Genevée C, Bosq J, Duvillard P, Avril MF, Hercend T (1993) Analysis of T cell receptor variability in tumor-infiltrating lymphocytes from a human regressive melanoma. Evidence for in situ T cell clonal expansion. J Clin Investig 91:1183–1190
Denkert C, Loibl S, Noske A, Roller M, Müller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Törne C, Weichert W, Engels K, Solbach C, Schrader I, Dietel M, von Minckwitz G (2010) Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 28:105–113
Gianni L, Zambetti M, Clark K, Baker J, Cronin M, Wu J, Mariani G, Rodriguez J, Carcangiu M, Watson D, Valagussa P, Rouzier R, Symmans WF, Ross JS, Hortobagyi GN, Pusztai L, Shak S (2005) Gene expression profiles in paraffin-embedded core biopsy tissue predict response to chemotherapy in women with locally advanced breast cancer. J Clin Oncol 23:7265–7277
Hornychova H, Melichar B, Tomsova M, Mergancova J, Urminska H, Ryska A (2008) Tumor-infiltrating lymphocytes predict response to neoadjuvant chemotherapy in patients with breast carcinoma. Cancer Invest 26:1024–1031
Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G (2008) The anticancer immune response: indispensable for therapeutic success? J Clin Investig 118:1991–2001
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira J-P, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, André F, Delaloge S, Tursz T, Kroemer G, Zitvogel L (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13:1050–1059
Ciampricotti M, Hau C-S, Doornebal CW, Jonkers J, de Visser KE (2012) Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med 18:344–346
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450:903–907
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman W-H, Pagès F, Galon J (2011) Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 71:1263–1271
Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie J–J, Rochaix P, Girard J-P (2011) Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res 71:5678–5687
Dieu-Nosjean M-C, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L, Lebecque S, Fridman W-H, Cadranel J (2008) Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 26:4410–4417
de Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, Cremer I, Fridman W-H, Sautès-Fridman C, Dieu-Nosjean M-C (2011) Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res 71:6391–6399
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307
Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, Scheppke L, Stockmann C, Johnson RS, Angle N, Cheresh D (2008) A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 456:809–813
Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS (2008) Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456:814–818
Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg S (2010) Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 70:6171–6180
Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O’Brien-Jenkins A, Gimotty P, Coukos G (2008) Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med 14:28–36
Dirkx AEM, Oude Egbrink MGA, Kuijpers MJE, van der Niet ST, Heijnen VVT, Bouma-ter Steege JCA, Wagstaff J, Griffioen AW (2003) Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res 63:2322–2329
Quezada S, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP (2008) Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med 205:2125–2138
Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, Rabie T, Kaden S, Gröne H-J, Hämmerling GJ, Arnold B, Ganss R (2008) Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 453:410–414
Jiménez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N (2000) Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med 6:41–48
Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, Fan AC, Yang Q, Braunstein L, Crosby E, Ryeom S, Felsher DW (2010) CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 18:485–498
Finlay D, Cantrell DA (2011) Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol 11:109–117
Onrust SV, Hartl PM, Rosen SD, Hanahan D (1996) Modulation of L-selectin ligand expression during an immune response accompanying tumorigenesis in transgenic mice. J Clin Investig 97:54–64
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM (2008) Batf3 deficiency reveals a critical role for CD8alpha + dendritic cells in cytotoxic T cell immunity. Science (New York, N.Y.) 322:1097–1100
Schrama D, thor Straten P, Fischer WH, McLellan AD, Bröcker EB, Reisfeld RA, Becker JC (2001) Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity 14:111–121
Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, Schietinger A, Philip M, Schreiber H, Fu Y-X (2004) Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol 5:141–149
Thompson ED, Enriquez HL, Fu Y-X, Engelhard VH (2010) Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med 207:1791–1804
Faget J, Biota C, Bachelot T, Gobert M, Treilleux I, Goutagny N, Durand I, Léon-Goddard S, Blay JY, Caux C, Ménétrier-Caux C (2011) Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res 71:6143–6152
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, Savino B, Colombo P, Jonjic N, Pecanic S, Lazzarato L, Fruttero R, Gasco A, Bronte V, Viola A (2011) Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med 208:1949–1962
Hong M, Puaux A-L, Huang C, Loumagne L, Tow C, Mackay C, Kato M, Prévost-Blondel A, Avril M-F, Nardin A, Abastado J-P (2011) Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res 71:6997–7009
Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HCJ, Haydon PG, Weninger W (2006) Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J Exp Med 203:2749–2761
Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S (2007) In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med 204:345–356
Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, Rüegg C, Dietrich P-Y, Walker PR (2005) Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 22:175–184
Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH (2003) Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 424:88–93
Mora JR, Cheng G, Picarella D, Briskin M, Buchanan N, von Andrian UH (2005) Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin- and gut-associated lymphoid tissues. J Exp Med 201:303–316
Mullins DW, Sheasley SL, Ream RM, Bullock TNJ, Fu Y-X, Engelhard VH (2003) Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med 198:1023–1034
Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT (2006) Regulation of lymph node vascular growth by dendritic cells. J Exp Med 203:1903–1913
Nakanishi Y, Lu B, Gerard C, Iwasaki A (2009) CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462:510–513
Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN (2006) Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 440:890–895
Kumamoto Y, Mattei LM, Sellers S, Payne GW, Iwasaki A (2011) CD4+ T cells support cytotoxic T lymphocyte priming by controlling lymph node input. Proc Nat Acad Sci USA 108:8749–8754
Wong SBJ, Bos R, Sherman LA (2008) Tumor-specific CD4+ T cells render the tumor environment permissive for infiltration by low-avidity CD8+ T cells. J Immunol (Baltimore, Md.: 1950) 180:3122–3131
Bos R, Sherman LA (2010) CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 70:8368–8377
Zou W, Restifo NP (2010) T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol 10:248–256
Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM (2007) IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol 8:369–377
Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C (2009) T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31:787–798
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114:1141–1149
Clark RA, Huang SJ, Murphy GF, Mollet IG, Hijnen D, Muthukuru M, Schanbacher CF, Edwards V, Miller DM, Kim JE, Lambert J, Kupper TS (2008) Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. J Exp Med 205:2221–2234
Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, Zhang B (2011) CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Investig 121:2371–2382
Cipponi A, Wieers G, van Baren N, Coulie PG (2011) Tumor-infiltrating lymphocytes: apparently good for melanoma patients. But why? Cancer Immunol Immunother 60:1153–1160
Umansky V, Sevko A (2012) Overcoming immunosuppression in the melanoma microenvironment induced by chronic inflammation. Cancer Immunol Immunother 61:275–282
Germeau C, Ma W, Schiavetti F, Lurquin C, Henry E, Vigneron N, Brasseur F, Lethé B, De Plaen E, Velu T, Boon T, Coulie PG (2005) High frequency of antitumor T cells in the blood of melanoma patients before and after vaccination with tumor antigens. J Exp Med 201:241–248
Acknowledgments
The authors thank Pierre Coulie, Oberdan Leo and Kris Thielemans for careful review and interesting suggestions. The Laboratory of Immunobiology is supported by grants of the Fonds National de la Recherche Scientifique (FNRS)/Télévie, by the Walloon Region (Programme d’excellence CIBLES). G.R. is supported by the Fondation Rose et Jean Hoguet.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rahir, G., Moser, M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol Immunother 61, 751–759 (2012). https://doi.org/10.1007/s00262-012-1253-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-012-1253-1