
Abstract
Severe immune suppression is frequent in late-stage tumor patients and promotes tumor immune evasion and subsequent tumor progression. Regulatory T cells (Treg) are major suppressors of anti-tumor immune responses. Therefore, targeting of Treg has become a key goal of anti-tumor therapy. Several preclinical and clinical observations suggest that Treg can be depleted by cyclophosphamide. Over a period of 3 months, we investigated the effect of metronomic low-dose cyclophosphamide on Treg numbers, suppressive capacity and proliferation on endogenous anti-tumor T-cell responses and on their correlation to clinical outcome in 12 patients with treatment-refractory metastasized breast cancer who received single-agent 50 mg cyclophosphamide p.o. daily. Cyclophosphamide treatment initially caused a significant reduction in circulating Treg by more than 40% (P = 0.002). However, Treg numbers completely recovered during the treatment due to increased proliferative activity and maintained their suppressive capacity. Treg depletion coincided with a strong increase in breast tumor–reactive T cells (P = 0.03) that remained at high levels during the whole period. Numbers of tumor-reactive T cells but not of Treg correlated with disease stabilization (P = 0.03) and overall survival (P = 0.027). We conclude that metronomic low-dose cyclophosphamide only transiently reduces Treg but induces stable tumor-specific T-cell responses, which correlate with improved clinical outcome in advanced-stage breast cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
CD4+CD25+Foxp3+ regulatory T cells (Treg) are key contributors to the prevention of autoimmune diseases as well as to the promotion of tumor progression [1–3]. Spontaneous anti-tumor T-cell responses are frequent in primary tumor patients [4] and their maintenance requires pertained exposure to tumor antigens [5], but in advanced-stage tumors, they are strongly inhibited by Treg at both stages of naïve T-cell priming [6, 7] and memory T-cell reactivation [8]. Treg accumulation in the tumors and draining lymph nodes of a variety of cancer entities [9–12] is associated with poor prognosis [10]. Given the detrimental role of Treg in tumor immunity, several clinical trials have aimed at their depletion through constitutively expressed surface markers [3]. However, circulating Treg were restored rapidly within 1 or 2 weeks after the depletion, while clinical responses were controversial.
Recently, cyclophosphamide (CTX) that belongs to a group of cytotoxic alkylating nitrogen mustard compounds was reported to have an effect on Treg reduction when applied solely at low dose and metronomic regimen in rodent models [13, 14]. In a study of several late-stage cancer patients, 30 days of metronomic low-dose CTX treatment selectively depleted CD4+CD25+Foxp3+ Treg [15], suggesting that prolonged treatment with low-dose CTX may not only control immune suppression by Treg but also support spontaneous anti-tumor T-cell responses and thereby clinical outcome of advanced-stage cancer patients.
To address these questions, we monitored for 3 months Treg frequencies and function, spontaneous anti-tumor T-cell responses as well as the clinical outcome in 12 treatment-refractory, progressively metastasized breast cancer patients undergoing treatment with 50 mg cyclophosphamide p.o. daily.
Materials and methods
Patients and treatment
Twelve patients were enrolled into this observational study approved by the Ethical Committee of the University of Heidelberg (Heidelberg, Germany). Informed consent was obtained from all participants. The mean age was 60.3 years (from 47 to 74 years). Eligibility criteria were as follows: stage IV breast cancers with measurable metastatic disease under progression after standard cytostatic therapy involving anthracyclines and taxanes; no secondary malignancies, autoimmune disease, renal failure or pregnancy (Table 1). Subjects received single-agent 50 mg cyclophosphamide (EndoxanR 50 mg, Baxter Oncology GmbH, Halle (Westf.), Germany) p.o. daily for 3 months. Assessments of toxicity were done at every clinic visit. Forty milliliters of peripheral blood (PB) was collected [16] before and fortnightly during the treatment. Immunological monitoring included analysis of Treg frequencies and numbers, of endogenous anti-tumor T-cell responses as assessed by ex vivo short-term IFN-γ ELISpot assays and by determination of in vivo T-cell proliferation and of the suppressive and proliferative activity of Treg. Clinical responses were monitored by quantification of tumor marker CA15-3 in patients’ sera, computed tomography (CT) or magnetic resonance tomography (MRT) of metastases when indicated (Table 1). Furthermore, overall survival was assessed in each patient.
As a control group, seven primary breast cancer patients (mean age, 57.3 years; range, 49–65) have been enrolled into this study. Informed consent was obtained from all participants. Eligibility criteria were as follows: female, no history of secondary malignancy and no metastatic disease. Additionally, as further controls, four healthy donors were included into the study after informed consent, all of them female without history of malignant disease (mean age, 56.8 years; range, 44–78).
Flow cytometry
A total of 5 × 105 ex vivo-isolated total PBMC or 1 × 105 ex vivo-purified CD4+CD25+ Treg were blocked with polyclonal human immunoglobulins (Endobulin, Baxter Oncology, Frankfurt, Germany; 2.5 mg/ml) for 15 min on ice. Cells were incubated with a combination of the following fluorochrome-conjugated monoclonal antibodies (mAb) (BD Pharmingen, Heidelberg, Germany): PerCP-Cy5.5-CD4 (clone SK3; dilution, 1:50), PE-CD3 (clone UCHT1; dilution, 1:50) and APC-Cy7-CD25 (clone 4E3; dilution, 1:25) for 20 min on ice. Intracellular APC-Foxp3 staining was conducted according to the manufacturer’s protocol (clone 236A/E7 eBioscience, San Diego, USA). Proliferation of different T-cell populations was assessed by costaining with FITC-anti-Ki67 mAb (clone B56; BD Pharmingen, Heidelberg, Germany; dilution, 1:10). Data acquisition and analysis were carried out using FACSCanto II (Becton–Dickinson, Heidelberg, Germany) and FlowJo Software (Tree Star, San Carlon, CA).
Preparation of T cells and dendritic cells for functional assays
T cells were cultured for 7 days in RPMI medium (PAA Laboratories, Cölbe, Germany) containing 10% AB serum (Sigma, Deisenhofen, Germany), 100 units/ml interleukin (IL)-2 (Chiron, Ratingen, Germany) and 60 units/ml IL-4 (Chiron, Ratingen, Germany). Afterward, T cells were transferred into cytokine-free medium for 12 h and subsequently separated from contaminating cells using T-cell negative isolation kit (Invitrogen, Karlsruhe, Germany). Treg were isolated in a two-step procedure according to their expression of CD4 and CD25 by magnetic beads using CD4+CD25+ regulatory T-cell isolation kit (Miltenyi, Bergisch Gladbach, Germany). Isolated CD4+CD25+ cells were collected, and the remaining T-cell populations (CD4−CD25+ and CD4+CD25−) were mixed and used as Treg-depleted T-cell fractions. Dendritic cells (DC) were generated from adherent monocytes according to standard procedures as described [16]. In brief, adherent cells from peripheral blood samples were cultured for 7 days in serum-free X-VIVO 20 (BioWhittaker, Vervier, Belgium) containing 50 ng/ml recombinant human granulocyte–macrophage colony-stimulating factor (Essex Pharma, München, Germany) and 1,000 units/ml IL-4. Afterward, differentiated DC were harvested from the supernatant and enriched using anti-CD3 mAb, anti-CD56 mAb and anti-CD19 mAb-coupled magnetic beads (Invitrogen, Karlsruhe, Germany) and pulsed for 14 h with 200 μg/ml of respective test or negative control antigens.
Antigens
Cell lysates from breast carcinoma cell lines MCF7 (ATCC #HTB-22, Rockville, USA) and KS24.22 (kind gift from B. Gückel, Department of Gynecology and Obstetrics, University of Tübingen, Tübingen, Germany) were generated through repeated freezing and thawing as described [16] and served as multivalent sources of breast tumor antigens, while a well-described synthetic long peptide from the breast cancer–associated antigen mucin-1 MUC1–100 was used as a defined tumor antigen. Lysate of the unrelated promonocytic leukemia cell line U937 (ATCC #CRL-1593.2, Rockville, USA) and a synthetic long peptide derived from the human IgG40–89 served as negative control antigens.
IFN-γ ELISpot assays
IFN-γ ELISpot assays were conducted as previously described [8]. Two hundred micrograms of respective antigen was used for pulsing of each 106 DC/ml over night. Antigen-loaded DC were incubated with autologous T cells in triplicate wells at a ratio of 1:10 for 40 h in ELISpot-plates (Millipore, Eschborn, Germany). IFN-γ spots were automatically measured using an Axioplan 2 imaging microscope and KS ELISpot software (Zeiss, Jena, Germany). Spots in negative control wells were considered as background. T cells were considered antigen-reactive when spot numbers in test wells exceeded significantly those in control wells (P < 0.05, Student’s t test). The frequency of tumor-reactive T cells was calculated as follows: (spots in test wells—spots in control wells)/T-cell numbers per well.
Treg suppression assay
Analysis of the functional capacity of Treg was conducted by the Treg Suppression Inspector Kit (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Treg were directly isolated from patient blood samples without in vitro passage. Thymidine incorporation by proliferating cells was measured using a scintillation counter (Liquid Scintillation Counter [1450 MicroBeta] Perkin Elmer, Wellesley, MA), as described [17].
Statistical analyses
Differences between mean values were calculated using two-sided Student’s t test. Enhancement in treatment-mediated tumor-specific T-cell responses was also assessed using Wilcoxon’s signed-rank test. Patients were considered as responders in ELISpot assays if spot numbers in the antigen test wells were statistically higher (P < 0.05) than those in negative control wells. Proportions of immune responders before and during treatment were compared using a two-sided Fisher’s exact test. Survival curves for different frequencies of tumor-reactive T cells were estimated, employing a fitted Cox regression model according to the observed survival data of study patients. The correlation between frequencies of tumor-reactive T cells and overall survival was assessed using Pearson’s product moment correlation. Threshold of significance was always set at P < 0.05.
Results
Patient treatment and analysis
Twelve metastasized breast cancer patients who were refractory to standard cytostatic treatment and under disease progression were subjected to treatment with 50 mg of cyclophosphamide p.o. daily (Table 1). Before and fortnightly until day 84 of treatment, peripheral blood (PB) was collected for immunological monitoring. Clinical responses were assessed by quantification of tumor marker CA15-3 and by computed tomography (CT) of metastases, when tumor marker levels were not representative (Table 1). The mean age was 60.3 years (from 47 to 74 years). CTX treatment was continued when patients showed stable disease after the study period of 3 months. Median treatment for stable patients has been 8 months (range, 5–13 months). CTX treatment was discontinued in patient #6 on day 14 due to tumor progression. Patient #4 resigned CTX treatment after 28 days despite partial remission of a brain metastasis. The treatment was well tolerated as none of the patients showed grade 3–4 side effects. We observed grade I leucopenia in all patients and anemia (hemoglobin 6.6 g/dl) in patient #6 requiring blood transfusions.
CTX treatment only transiently reduces Treg in peripheral blood
The frequency of CD4+CD25+Foxp3+ Treg among total CD4+ T cells was assessed by flow cytometry (Fig. 1a). Before CTX treatment, Treg were slightly but not significantly increased in patients compared to healthy individuals (Fig. 1b). Within 14 days of CTX treatment, Treg were consistently reduced in all patients (3.0 ± 1.8% versus 5.1 ± 3.0%; P = 0.002) and remained decreased until day 42 (Fig. 1c). Afterward, Treg gradually recovered until their complete restoration was observed on day 84 of CTX treatment (5.2 ± 2.1%). Similarly, absolute numbers of Treg were reduced within 14 days (from 19,000 ± 8,600/ml to 11,000 ± 1,200/ml) and gradually increased subsequently until pre-treatment levels were reached on day 56 (22,000 ± 7,700/ml), which was not the case with absolute numbers of CD4+ T cells (data not shown).

Impact of CTX treatment on Treg frequency in the blood. a Definition of CD4+CD25highFoxp3+ Treg by flow cytometric assay. b Comparison of Treg frequencies in healthy donors and advanced breast cancer patients. Column+bar value+SEM. HD healthy donors, Adv. Pat. advanced breast cancer patients, n number of assessed samples. c Treg frequencies during CTX treatment. Individual values are depicted by dots and connected by lines. Patient numbers are marked on Day 0 nearby corresponding dots. Horizontal bars, mean values at the time indicated
These data demonstrate that metronomic low-dose cyclophosphamide treatment selectively reduced the number of Treg for 4–6 weeks but was insufficient to maintain Treg at low frequencies for longer periods.
CTX treatment induces stable anti-tumor T-cell responses
In order to test whether CTX treatment affects endogenous T-cell responses against breast tumor–derived antigens, we quantified ex vivo-isolated tumor-reactive T cells fortnightly throughout the study by short-term (40 h) IFN-γ ELISpot assays. As source of breast tumor–derived antigens, we used lysates of the well-defined breast cancer cell lines MCF7 [18, 19] and KS24.22 [20], which together express a broad variety of breast cancer–associated antigens, while lysates of the promonocytic leukemia cell line U937 as a source of unrelated tumor antigens served as negative control antigens.
Figure 2a shows results obtained from one representative patient who developed breast tumor–reactive T cells within 14 days of treatment. Tumor-reactive T cells were then detectable throughout the full study. Within the total study group, we detected breast tumor–reactive T cells in 27% of the patients already before CTX treatment (Fig. 2b). During treatment, this proportion was substantially increased up to 73% (P = 0.086) on day 14 and maintained at high levels throughout the entire study with 80% on day 42 (P = 0.03) and 88% on day 84 (P = 0.02, Fig. 2b).

Tumor-specific T cells during CTX treatment. Exemplary (a) and cumulative (c) results of IFN-γ ELISpot analyses. Column+bar mean value+SEM. Triangles individual patients. b Proportions of tumor responding patients. Patients were considered as responders if they contained statistically higher (P < 0.05) MCF7 or KS24.22-reactive T cells than U937-reactive ones. The differences before and during the treatment were compared using a two-sided Fisher’s exact test. n number of responders/total number of patients assessed. d Unmasking of tumor-reactive T cells by Treg depletion in patients 2, 12 and 9 (left panel) but not in patients 4, 6 and 8 (right panel). *P < 0.05; **P < 0.005; ***P < 0.0005
The calculated frequencies of breast tumor–reactive T cells among total T cells are shown in Fig. 2c. During the treatment period, their frequency increased gradually by more than threefold from 12 ± 8.2/105 T cells initially until a significant increase up to 39 ± 9.7/105 T cells was detected in the last month of treatment (P = 0.04). Taken together, these results provide evidence that metronomic low-dose cyclophosphamide treatment stably increases breast tumor–reactive T-cell responses despite an only transient effect on Treg.
The increased T-cell responses might be either due to a reduced suppression of pre-existing tumor-specific T cells or due to an increased de novo induction of specific T-cell responses. In order to address this issue, we evaluated in 6 untreated study patients whether or not pre-existing Treg suppressed the activity of pre-existing tumor-specific T cells (Fig. 2d). To this end, we depleted their isolated T cells of Treg and assessed subsequently T-cell reactivity against breast tumor–derived antigens (patients #2, 4, 6, 8, 12) or against the defined tumor antigen MUC-1 (patient #9) by IFN-γ ELISpot analysis. In three patients (patients #2, 9, 12), breast tumor–specific T-cell responses were revealed (patient #2) or increased (patients #9, 12) after depletion of Treg. Interestingly, in patient #2, the anti-tumor T-cell responsiveness as induced by Treg depletion was similar to observed frequencies after 42 days of CTX treatment. In the other three patients (patients #4, 6, 8), even after depletion of Treg, we did not detect breast tumor–reactive T cells. However, strong breast tumor–reactive T-cell responses were detectable during CTX treatment. These observations suggest that CTX treatment can reveal pre-existing but suppressed T-cell responses as well as trigger a de novo generation of tumor-specific T cells.
CTX treatment-induced increase in breast tumor–reactive T cells correlates with induction of stable disease and with enhanced overall survival
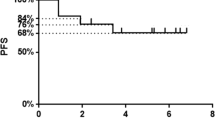
While all patients were in progressive disease (PD) before treatment, stable disease (SD) was achieved in 7 out of 12 (58%) patients according to their development of tumor markers and/or CT/MRT assessment (Table 1). Five patients (42%) remained in PD. Clinical outcome did not correlate to grading, hormone receptor expression, proliferation index (Ki-67) and Her2/neu status of the primary tumor (data not shown). During CTX treatment, in the group of patients achieving SD, however, we observed a significant increase in breast tumor–reactive T cells compared to pre-treatment levels (P = 0.03, Fig. 3a).

Correlation of breast tumor–specific T-cell reactivity with disease status and overall survival. a CTX treatment-mediated elevation of breast tumor–specific T cells is significantly associated with disease stabilization. Column+bar mean value+SEM. PD progressive disease, SD stable disease. b Detected frequencies of breast tumor–reactive T cells at the end of CTX treatment is significantly correlated to overall survival. Each dot represents one assessed patient
We analyzed overall survival (OS) for a period of up to 76 weeks after the initiation of CTX treatment. Within this observation period, 8 patients died of tumor progression, while 4 patients are still alive (Table 1). Interestingly, after CTX treatment, we detected a correlation between the number of breast tumor–reactive T cells and OS (P = 0.027, Fig. 3b). In contrast, the frequencies of Treg during or after treatment had no influence on SD or OS (data not shown).
CTX treatment does not affect Treg function but increases the expansion of CD4+ and CD8+ Tconv and Teff cells
In view of the observed discrepancy between a complete restoration of the Treg pool under treatment and a simultaneous increase in the frequencies of breast tumor–reactive T cells, we wondered whether CTX treatment might inhibit Treg function while leaving expansion of the Treg pool unaffected. In order to address this, we purified Treg (Fig. 4a) from the blood of patients on day 84 or from healthy individuals and tested their capacity to suppress the proliferation of autologous conventional T cells (Tconv) after polyclonal T-cell stimulation. The original results obtained from one exemplary study patient are shown in Fig. 4b, demonstrating a substantial, dose-dependent suppression of Tconv proliferation by Treg. Interestingly, as demonstrated in Fig. 4c, we did not detect any differences in the suppressive capacity of Treg from healthy individuals and CTX-treated study patients. Thus, restored Treg in CTX-treated patients possess an unaltered suppressive capacity.

Treg on day 84 of CTX treatment maintain suppressive capacity. a Purity of enriched Treg measured by flow cytometric analysis. Cells were pre-gated on CD3+CD4+ T cells. Small number shown in the rectangle represents the percentage of CD25+Foxp3+ cells among CD4 T cells. b Exemplary results of Tconv proliferation assay. Column+bar mean value+SEM. c Cumulative data of suppressive potential of Treg isolated from healthy donors (HD) or study patients shown as % inhibition of Tconv proliferation. Horizontal bars means. n number of assessed subjects, Treg regulatory T cells, Tconv conventional T cells
The observed replenishment of the Treg pool might be explained by increased de novo generation (e.g., through conversion of Tconv) [21] or by increased proliferation of Treg. In order to assess the latter possibility, we analyzed by flow cytometry the expression of the intracellular proliferation marker Ki67 in a variety of T-cell populations, involving CD25+Foxp3+CD4+ or CD8+ regulatory T cells under CTX treatment. The data were compared to respective T cells from untreated primary breast cancer patients and healthy donors (Fig. 5).

Proliferative capacity of different T-cell subsets during CTX treatment. Frequency of Ki67-expressing CD4+ (left panel) and CD8+ (right panel) T cells. T cells were subgrouped into Treg (a) and Teff (b) according to CD25 and Foxp3 expression. Healthy donors (HD) (filled black circle), primary breast cancer patients (Pri. BrCa) (filled blue circle) and study patients on day 0 (open blue circle), day 56 (filled blue triangle), day 70 (open blue triangle) and day 84 (filled blue diamond). Symbols, individual assessments. Horizontal bars, median values. *P < 0.05
With regard to proliferating CD4+ Treg (Fig. 5a, left), healthy individuals showed a median proportion of 11.9% of total Treg, most likely indicating the degree of homeostatic proliferation under normal conditions. A slightly higher proportion (14.8%) were observed in primary breast cancer patients, while Treg proliferation was strongly increased in advanced breast cancer patients (22.45%, Day 0), indicating a stage-associated abnormal amplification of Treg. Treg proliferation in CTX-treated patients was reduced on day 56 (16.9%) compared to day 0 but gradually increased to pre-treatment levels until day 84 (21.0%). These data indicate that at late study points of time, the proliferative capacity of CD4+ Treg fully recovered despite ongoing CTX treatment. Interestingly, in the subpopulation of CD8+CD25+Foxp3+ Treg, we even observed a significantly increased proportion of proliferating cells on day 84 when compared to those of healthy donors, primary breast cancer patients or study subjects on day 0 (Fig. 5a, right). Thus, replenishment of Treg under CTX treatment can at least partially be explained by increased proliferation.
In CD4+ and CD8+ effector T cells (Teff), reduced proliferation proportions were observed in both primary and untreated advanced breast cancer patients as compared to healthy donors (Fig. 5b, left and right). Interestingly, however, proliferating abilities of Teff were normalized or even further enhanced under CTX treatment. Notably, this effect was stronger in CD8+, potentially cytotoxic T cells (Fig. 5b, right), which might have contributed to the improved clinical outcome as observed in the present study.
Discussion
Since the first clinical trial of metronomic chemotherapy involving cyclophosphamide in 2002 [22], such low-dose cyclophosphamide-containing regimens demonstrated anti-tumor efficacy with minimal toxicity in advanced cancer patients [23]. Preclinical mouse models and a previous clinical study on a number of advanced cancer patients suggested that the anti-tumor efficacy of low-dose cyclophosphamide is linked to a reduction and functional impairment of Treg, which might improve immune responsiveness to subsequent tumor vaccination [13–15, 24]. However, effects of such treatment on the stimulation of endogenous immune responses have not been evaluated in patients with cancer. This concern prompted us to perform the present study in patients with metastatic advanced breast cancer. We here show that single application of metronomic low-dose cyclophosphamide over 3 months induced breast tumor–specific T-cell responses in most of the patients. There is ever-expanding evidence that chemotherapy exhibits superior therapeutic effects when the host is immunocompetent [25]. In accordance with these findings, we observed that disease stabilization and overall survival correlated with the extent of anti-tumor T-cell responsiveness.
Spontaneous anti-tumor T-cell responses against a relatively wide scope of tumor-associated antigens (TAA) have been described previously in primary breast cancer [26]. Evidence suggests that the breadth and diversity of T-cell responses are crucial for cancer immunity [27]. On this basis, we monitored the frequencies of tumor-specific T cells against a broad repertoire of breast TAAs contained in breast cancer cell lines MCF7 [18, 19] and KS24.22 [20]. As confirmed with both antigen sources, CTX treatment was able to substantially elevate the proportion of patients with breast tumor–reactive T cells and their frequencies within 14 days and to maintain these for the whole observation period.
Several mechanisms may contribute to the immunological effects of this treatment. Firstly, due to its anti-angiogenic and cytotoxic activity [28–30], this regimen might provide the advantage that more TAAs become available for T-cell priming. Secondly, as indicated by previous experimental results [14, 31] and data from late-stage cancer patients [15], such treatment induces a selective deprivation of immunosuppressive Treg but preserves the proliferative activity of conventional T cells (Tconv). The observed selective sensitivity of Treg to cyclophosphamide treatment may be explained, at least partially, by their constitutively reduced content of ATP [31], which nevertheless does not explain their rapid restoration during continued cyclophosphamide treatment.
In line with the aforementioned findings, we report here that metronomic cyclophosphamide treatment resulted in a selective and rapid although only transient decrease in Treg numbers. This transient withdrawal of immunosuppressive influences may have contributed to the observed unmasking of pre-existing memory T cells as well as to the subsequent de novo generation of breast tumor–specific effector T-cell responses. Recently, it was demonstrated that cyclophosphamide treatment in mice increased T-cell immunity through a selective depletion of CD8+ DC together with an increased turnover, intratumoral accumulation and IL-12 secretion of migratory DC subsets [32, 33]. It is of interest to note that the treatment-induced proliferation was observed in both CD4 and CD8 subsets, but more dramatically within the latter subset of potentially cytotoxic T lymphocytes.
One pertinent question in this study concerns the impact of long-term CTX regimens on Treg. Under this treatment, the pool of CD4 Treg was unexpectedly replenished. Moreover, in contrast to a report on patients treated with low-dose cyclophosphamide for only 30 days resulting in a strong functional impairment of Treg [15], we did not observe any alteration of the suppressive function of Treg after 84 days of this treatment. Both phenomena might be attributed to homeostatic proliferation or to a possible conversion from Tconv [21]. Although we cannot exclude the latter possibility, the increased proliferative capacity of the Treg population as determined by Ki67 expression supports the former notion. In this context, the concomitantly proliferating CD4 Tconv may serve as a source of IL-2, which is an essentially required cytokine for Treg expansion and survival [34, 35].
Last but not least, Treg play an important role in the maintenance of peripheral tolerance against autoantigens, and their depletion causes severe autoimmune disorders. This might be an obstacle of short-term cyclophosphamide application, which in clinical studies exerted profound functional and quantitative deprivation of Treg [36, 37]. In this regard, it may be of clinical relevance that the long-term administration of cyclophosphamide, as conducted in our present study, allowed for their functional and quantitative restoration, while the pool of restored Treg apparently no longer suppressed tumor-specific type-I T-cell responses.
References
Sakaguchi S (2005) Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol 6:345–352
Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, Kuniyasu Y, Nomura T, Toda M, Takahashi T (2001) Immunologic tolerance maintained by CD25+CD4+ regulatory T cells: their common role in controlling autoimmunity, tumour immunity and transplantation tolerance. Immunol Rev 182:18–32
Zou W (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6:295–307
Solomayer EF, Feuerer M, Bai L, Umansky V, Beckhove P, Meyberg GC, Bastert G, Schirrmacher V, Diel IJ (2003) Influence of adjuvant hormone therapy and chemotherapy on the immune system analysed in the bone marrow of patients with breast cancer. Clin Cancer Res 9:174–180
Mahnke YD, Schwendemann J, Beckhove P, Schirrmacher V (2005) Maintenance of long-term tumour-specific T-cell memory by residual dormant tumour cells. Immunology 115:325–336
Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, Niesner U, de la Rosa M, Schmidt CA, Bräuer R, Buer J, Scheffold A, Hamann A (2004) Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med 199:303–313
Liu Z, Kim JH, Falo LD Jr, You Z (2009) Tumor regulatory T cells potently abrogate antitumour immunity. J Immunol 182:6160–6167
Bonertz A, Weitz J, Pietsch DH, Rahbari NN, Schlude C, Ge Y, Juenger S, Vlodavsky I, Khazaie K, Jaeger D, Reissfelder C, Antolovic D, Aigner M, Koch M, Beckhove P (2009) Antigen-specific Tregs control T-cell responses against a limited repertoire of tumour antigens in patients with colorectal carcinoma. J Clin Invest 119:3311–3321
Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC (2002) Prevalence of regulatory T cells is increased in peripheral blood and tumour microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 169:2756–2761
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Erdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zim Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH (2001) Regulatory CD4(+)CD25(+) T cells in tumours from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res 61:4766–4772
Viguier M, Lemaître F, Verola O, Cho MS, Gorochov G, Dubertret L, Bachelez H, Konrilsky P, Ferradini L (2004) Foxp3 expressing CD4+ CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol 173:1444–1453
Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F (2004) CD4+CD25+ regulatory T cells suppress tumour immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumours to be curative. Eur J Immunol 34:336–344
Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H (2005) Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105:2862–2868
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B (2007) Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 56:641–648
Feuerer M, Beckhove P, Bai L, Solomayer EF, Bastert G, Diel IJ, Pedain C, Oberniedermayr M, Schirrmacher V, Umansky V (2001) Therapy of human tumours in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat Med 7:452–458
Nummer D, Suri-Payer E, Schmitz-Winnenthal H, Bonertz A, Galindo L, Antolovich D, Koch M, Büchler M, Weitz J, Schirrmacher V, Beckhove P (2007) Role of tumour endothelium in CD4+CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst 99:1188–1199
Soule HD, Vazguez J, Long A, Albert S, Brennan M (1973) A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst 51:1409–1416
Kämmerer U, Thanner F, Kapp M, Dietl J, Sütterlin M (2003) Expression of tumour markers on breast and ovarian cancer cell lines. Anticancer Res 23:1051–1055
Gückel B, Stumm S, Rentzsch C, Marmé A, Mannhardt G, Wallwiener D (2005) A CD80-transfected human breast cancer cell variant induces HER-2/neu-specific T cells in HLA-A*02-matched situations in vitro as well as in vivo. Cancer Immunol Immunother 54:129–140
Vignali DA, Collison LW, Workman CJ (2008) How regulatory T cells work. Nat Rev Immunol 8:523–532
Colleoni M, Rocca A, Sandri MT, Zorzino L, Masci G, Nolè F, Peruzzotti G, Robertson C, Orlando L, Cinieri S, Viale G, Goldhirsch A (2002) Low-dose oral methotrexate and cyclophosphamide in metastatic breast cancer: antitumour activity and correlation with vascular endothelial growth factor levels. Ann Oncol 13:73–80
Scharovsky OG, Mainetti LE, Rozados VR (2009) Metronomic chemotherapy: changing the paradigm that more is better. Curr Oncol 16:7–15
Correale P, Cusi MG, Tsang KY, Del Vecchio MT, Marsili S, Placa ML, Intrivici L, Aquino A, Micheli L, Nencini C, Ferrari F, Giorgi G, Bonmassar E, Francini G (2005) Chemo-immunotherapy of metastatic colorectal carcinoma with gemcitabine plus FOLFOX 4 followed by subcutaneous granulocyte macrophage colony-stimulating factor and interleukin-2 induces strong immunologic and antitumour activity in metastatic colon cancer patients. J Clin Oncol 23:8950–8958
Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G (2008) The anticancer immune response: indispensable for therapeutic success? J Clin Invest 118:1991–2001
Sommerfeldt N, Schütz F, Sohn C, Förster J, Schirrmacher V, Beckhove P (2006) The shaping of a polyvalent and highly individual T-cell repertoire in the bone marrow of breast cancer patients. Cancer Res 66:8258–8265
Messaoudi I, Guevara Patin JA, Dyall R, LeMaoult J, Nikolich-Zugich J (2002) Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science 298:1797–1800
Dellapasqua S, Bertolini F, Baqnardi V, Campagnoli E, Scarano E, Torrisi R, Shaked Y, Mancuso P, Goldhirsch A, Rocca A, Pietri E, Colleoni M (2008) Metronomic cyclophosphamide and capecitabine combined with bevacizumab in advanced breast cancer. J Clin Oncol 26:4899–4905
Bocci G, Nicolaou KC, Kerbel RS (2002) Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res 62:6938–6943
Shaked Y, Emmenegger U, Man S, Cervi D, Bertolini F, Ben-David Y, Kerbel RS (2005) Optimal biologic dose of metronomic chemotherapy regimens is associated with maximum antiangiogenic activity. Blood 106:3058–3061
Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B (2010) Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res 70:4850–4858
Nakahara T, Uchi H, Lesokhin AM, Avogadri F, Rizzuto GA, Hirschhorn-Cymerman D, Panageas KS, Merghoub T, Wolchok JD, Houghton AN (2010) Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood 115:4384–4392
Radojcic V, Bezak KB, Skarica M, Pletneva MA, Yoshimura K, Schulick RD, Luznik L (2010) Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol Immunother 59:137–148
Malek TR, Yu A, Vincek V, Scibelli P, Kong L (2002) CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity 17:167–178
Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ (2002) Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J Exp Med 196:851–857
Sakaguchi S (2000) Regulatory T cells: key controllers of immunologic self-tolerance. Cell 101:455–458
Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, Duray PH, Steinberg SM, Allison JP, Davis TA, Rosenberg SA (2003) Cancer regression and autoimmunity induced by cytotoxic T lymphocyte associated antigen-4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA 100:8372–8377
Acknowledgments
No grants or financial support were received by any of the authors in relation to this study or the writing of this article.
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Ge and C. Domschke contributed equally.
Rights and permissions
About this article
Cite this article
Ge, Y., Domschke, C., Stoiber, N. et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother 61, 353–362 (2012). https://doi.org/10.1007/s00262-011-1106-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-011-1106-3