
Abstract
Lung carcinoma is one of the leading causes of death worldwide. It is a non-immunogenic cancer, resistant to immune surveillance. Toll-like receptors (TLRs) connect the innate to the adaptive immune system. Given that cancerous cells evade the immune system, the activation of TLRs could represent a potential target for cancer therapy. The induction of Th1-like and cytotoxic immunity by TLR signalling could lead to tumour cell death, resulting in tumour regression or arrest. However, basic research and clinical trials revealed that the activation of specific TLRs, such as TLR2, TLR4 and TLR9, do not have any anti-tumour activity in lung carcinoma. Increasing evidence suggests that TLRs are important regulators of tumour biology; however, little is known about their function in lung cancer. Thus, in order to develop new therapeutic approaches, further studies are needed to understand the connection between TLRs and lung cancer progression. This review focuses on the potential mechanisms by which TLR ligands can facilitate or not lung cancer and lung metastases establishment/progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung carcinoma is one of the leading causes of death worldwide. Despite advances in treatment, the prognosis remains poor, with only 15% of patients surviving more than 5 years from the time of diagnosis. It is a non-immunogenic cancer, resistant to the surveillance of the immune system [1, 2], which should protect the body from tumour development by recognizing cancerous cells. The process of shaping the immunogenicity of tumours has been termed ‘cancer immune-editing’, which relies upon the activation of the adaptive immune system to recognize and eliminate ‘transformed’ cells [3]. Therefore, the activation of the immune system could represent a means to induce tumour regression [2, 3]. However, many studies have associated inflammation, thus the activation of the immune system, to cancer [4]. The tumour microenvironment is characterized by a process of chronic inflammation, which can facilitate either tumour establishment or progression [4].
Toll-like Receptors (TLRs) play a key role in the innate immune system. The recognition of specific molecules by TLRs leads to the activation of the adaptive immune system. Thus, the exogenous activation of the immune system via TLRs could on one side be a strategy to fight cancer, but on the other exacerbate, a latent chronic inflammation, which in turn, would further favour lung carcinoma progression.
This review focuses on the differential effects of TLR activation in lung carcinoma and explains the inefficiency of most TLR ligands as anti-cancer adjuvants in lung carcinoma therapy, although they were described as potential adjuvants for anti-cancer vaccines [5].
Toll-like receptors and their ligands
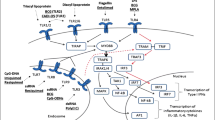
Toll-like Receptors (TLRs) recognize conserved molecular patterns in microbes (pathogen-associated molecular patterns: PAMPs) and endogenous ligands (danger-associated molecular patterns: DAMPs) [6]. TLRs are expressed either on the plasma membrane or in endosomal/lysosomal organelles. Cell surface TLRs recognize the lipopolysaccharide of Gram negative bacteria (TLR4), bacterial lipoproteins (TLR2/1 and TLR2/6) and flagellin (TLR5); whereas endosomal TLRs mainly recognize nucleic acids, such as double-stranded RNA (TLR3), single stranded RNA (TLR7) and double stranded DNA (TLR9) [4, 7] (Fig. 1). The activation of all TLRs leads to myeloid differentiation factor 88 (MyD88)-dependent signalling pathway, except for TLR3, which signals in a TIR-domain-containing adapter-inducing interferon-β (TRIF)-dependent manner (Fig. 1). TLR4 is the only receptor that can induce both MyD88- and TRIF-dependent signalling pathways [6]. MyD88- or TRIF-dependent signalling pathways induce the synthesis or release of pro- and anti-inflammatory cytokines and chemokines via the activation of transcription factors, such as NF-κB, interferon regulatory factor (IRF)-3/7, AP-1 and others [7, 8] (Fig. 1).

TLR signalling and their ligands
Lung TLRs in health and cancer
TLRs are widely expressed on both resident lung cells as well as on infiltrating cells of myeloid and lymphoid origin. Airway epithelial cells express high levels of TLR1, TLR2, TLR3, TLR4, TLR5, TLR6 and TLR9 [7, 9, 19]. Interestingly, TLR2, TLR4, TLR7/8 and TLR9 are over-expressed in lung carcinoma compared with the ‘normal’ lung [11, 12]. Similarly, TLRs are expressed on the infiltrating immune cells, which activation can dictate the adaptive immunity polarization.
The fine interaction between the stromal and the haematopoietic cell lineage in the lung becomes very important during cancerous conditions. The activation of TLRs on both cell types can facilitate the recruitment of immuno-stimulant or tolerogenic cells to the lung, where they can participate in the oncogenic process. The activation of TLRs on epithelial cells promotes the production of chemokines, such as CXCL-8 (IL-8), a potent neutrophil chemoattractant [9], and the release of growth factors, such as vascular endothelium-derived growth factor (VEGF) that recruits blood vessels to inflamed and damaged airway epithelium [13]. Similarly, the activation of TLRs on innate immune cells, such as Antigen-Presenting Cells (APCs) (i.e. Dendritic Cells (DCs), can enhance the adaptive immune system by the following mechanisms: (i) antigen processing and presentation, (ii) up-regulation of co-stimulatory molecules, such as CD80/86, which are necessary for CD4+ T-cell activation from naïve CD4 T cells [14] and(iii) modulation of T regulatory cells (Treg) activity by the production of some cytokines such as IL-6 [15]. TLR-induced synthesis/release of pro-inflammatory cytokines can dictate the adaptive immunity polarization. For example, TLR ligation can lead to the production of IL-12p35 or IL-12p19 and IL-27, which promote Th1 or Th17 immunity, respectively [16]. A Th1-like bias can affect the immune surveillance to an effective anti-tumour immune response in lung carcinoma (Fig. 2). In contrast, TLR-mediated Th17 immunity can exert either pro-tumour [17] or anti-tumour [18] activity depending on the mouse tumour model and on the tissue/organ. An outstanding study on IL-17 knockout mice demonstrated that the Th17 immunity has anti-tumour activity in lung carcinoma [18].

The activation of Antigen Presenting Cells (APCs), such as DCs, mast cells and B cells, via TLRs can lead to differential adaptive immunity. The induction of a Th1-like as well as T cytotoxic (Tc)-like immunity leads to tumour regression, differently than a Th2-like and T reg-mediated immunity
It seems clear that TLR-mediated synthesis/release of inflammatory mediators from lung stromal and immune cells dictates pulmonary immune microenvironment paving the way for lung cancer establishment/progression [10]. In particular, the insult of an inert antigen [10] or low concentrations of an antigen [11] can sensitize and facilitate a Th2-like immunity that, in contrast to a Th1-like immunity, dampens the host response, favouring a latent chronic inflammation. The progression of chronic alterations in the pulmonary network can be at the basis of tumour establishment/progression in the lung [4, 8]. The predominance of a Th2-like chronic inflammation in the lung can facilitate the proliferation of ‘transformed’ cells, which are not recognized by the immune system. Moreover, it is to note that immunosuppressive networks and especially T regulatory (Treg) cells can alter the Th2-like response facilitating the chronic inflammation and thus tumour immune escape in the lung [2, 3] (Fig. 2).
It is thus clear that the activation of TLR-dependent signalling pathways can play a fundamental role in tumour immunology, deciding for the fate of cancerous cells in the lung. Increasing evidence has highlighted the involvement of TLRs in the progression and development of cancer [8, 22]. In the nineteenth century, the involvement of TLRs in cancer was firstly observed by William Coley who discovered that repeated injections of bacterial toxins (later identified as LPS-Coley’s toxin) could serve as an efficient anti-tumour adjuvant [1]. Since then, numerous studies have been conducted on TLR ligands and their anti-tumour activity. However, the debate on the role of TLRs in cancer is still open, since they can either participate to the activation of the immune system to fight cancerous cells or promote carcinogenesis by the enhancement of chronic inflammatory responses [4, 7, 8, 22].
Activation of TLRs on dendritic cells (DCs) in lung cancer
DCs bridge the innate to the adaptive immune system. They line the external microenvironment and lung epithelium, where they survey the tissues for incoming antigens [23]. Lung DCs can be simply divided into conventional and plasmacytoid DCs (pDCs) [24]. When immune homoeostasis of the lung is disturbed by an inciting pathogen or inflammatory insult, additional monocyte-derived DCs are rapidly recruited to the lung, where they cooperate with the resident cDCs [23]. DCs express all the receptors of the innate immune system, such as TLRs, and have the potential to take up the antigen, process it and present it to T cells, so that to evoke an adaptive immune response. The acquisition of antigens by lung DCs is TLR-dependent. Human and mouse myeloid DCs (mDCs) express TLR1, 2, 3, 4, 5, 6, 7/8 and 9 [25, 28], which can modulate DC activation markers [14, 26]. However, the role of all TLRs on DCs in lung carcinoma still needs elucidation. In the absence of TLR ligation, lung DCs are minimally active [23]. The activation of TLR2 and TLR4 on lung-derived DCs can lead to the proliferation of either Th1- or Th2-like immune responses [21]. This dichotomy maybe explained by the dose of PAMPs inhaled and recognized by the specific TLR. An example is LPS, the TLR4 ligand, that at high doses can induce a Th1-mediated response, but at low doses, it facilitates a Th2-like immune response in a MyD88-dependent manner in the lung [21]. The adoptive transfer into the lung of an innocuous pathogen-pulsed DCs can facilitate a Th2 skew leading to immune tolerance [21]. Lung cancer is characterized by a Th2-like microenvironment, where DCs, that highly populate the tumour microenvironment, are in their immature state [22, 25, 26]. Immature DCs present lower expression of activation markers, such as CD80/86 [14] and MHC II [26]. It is likely that immature mDCs in lung carcinoma could be due to the presence of pDCs, as described in Th2-based lung pathologies, such as asthma [29], and some viral [30] and bacterial infections [26]. Our published data show that the ablation of pDCs facilitates mDCs to their mature phenotype associated with higher presence of the cytotoxic CD8+ T cells and less Treg, resulting in tumour arrest in a mouse model of lung carcinoma [22]. However, the molecular mechanism by which pDCs render mDCs immature is still not known. We could speculate that the expression of tolerogenic patterns in pDCs can modify mDCs activity in an indirect or cell–cell contact manner. Nonetheless, it is conceivable that mDCs are rendered immature by tolerogenic factors (i.e. TGFβ and IDO metabolites) produced by stromal cells in a TLR-dependent manner.
pDCs uniquely and highly express TLR7/8 and TLR9 [28]. The activation of these receptors leads to the production of IFNα/β in a MyD88-dependent manner [31]. IFN type I was primarily considered beneficial for tumour immuno-editing, acting on the homoeostasis of haematopoietic cells [25, 31, 32], since it can stimulate NK cell proliferation and activation, CD8+ T-cell cytotoxicity, cytokine secretion, TRAIL-mediated cell death and cross-presentation [32]. However, host targets of IFN type I maybe multiple and its function has not been clearly defined yet in lung carcinoma. Since pDCs are the major IFN-producing cells, it is likely that the tumour microenvironment disables their anti-tumour function [33]. Indeed, the high production of lung prostaglandin E2 (PGE2) and TGFβ, an immunosuppressive cytokine, reduces the pro-inflammatory activity of pDCs, facilitating the tolerogenic phenotype enforced by the direct pDC-mediated recruitment of Tregs [34, 35]. There is no direct evidence about PGE2, IFNα/β and pDCs in lung cancer models; however, it might be the reason of that pDCs are tolerogenic in the lungs of cancer patients [22, 26]. It is of note that the differential microenvironment in tumours can dictate the immune cell phenotype and that the choice of the experimental mouse model is fundamental to understand the immune microenvironment in lung carcinoma. Indeed, several studies on lung carcinoma are based on the implantation of tumour cells on the right flank of mice; however, this mouse model does not mimic the pulmonary environment. Moreover, transgenic mouse models for Her2, epithelial growth factor receptor (EGFR) and kRas are not appropriate for the study of tumour immune escape and even less for the implication of TLRs in lung carcinoma, since the ligation of some TLRs induces the activation of Her2, EGFR and kRas-dependent signalling pathways [13, 19, 31].
Moreover, the activation of TLR2, TLR4, TLR7/8 and TLR9, all of which are highly expressed in lung carcinoma tissues, induce the production of IFN type I and IFNγ that in turn can facilitate the expression of some tolerogenic enzymes, such as indoleamine-2, 3-dyoxigenase (IDO) [28, 30], and immunosuppressive cytokines, such as IL-10 and TGFβ [30], that are implicated in the generation of adaptive and natural Tregs, respectively [37]. pDCs can also directly bind to Treg via PD-1/PD-L1 axis, enhancing the tolerogenic scenario in lung cancer [34, 36]. Moreover, the production of TGFβ can lead to either innate Treg or Th17 generation [35]. TLR4, 7 and 9 activations promote TGFβ release, which in the presence of IL-6, an NF-κB-dependent cytokine [37], leads to a predominant Th17 immune response rather than Treg-mediated tolerance [37]. The genetic absence of IL-17 increases lung metastasis under TLR9 stimulation with CpG-ODN [18]; however, other tumour animal models show reduced tumour burden in IL-17 receptor (IL-17R) knockout mice [17] The discrepancy on the role of IL-17 in cancer could be due to the knockout animal models or to the tissue-specificity.
Activation of TLRs on B cells in lung cancer
TLRs are crucial for the humoral immune system orchestrated by B cells, which undergo maturation, proliferation and immunoglobulin switching following TLR activation [34]. The number of B cells in the lung during physiological conditions is very low (≈0.1%), but it is threefold increased during lung cancer, especially in the absence of tolerogenic cells [our unpublished data and 40]. Controversial studies refer to B cells as both anti-tumour and pro-tumour cells. B cells express high levels of TLR7 and TLR9 [28]. Some authors assumed that B cells countered T-cell-mediated immunity enhancing tumour cell growth [41]. The pro-tumour activity of B cells was ascribed to their regulatory phenotype. In sharp contrast, our published data show that CpG, TLR9 ligand, polarizes B cells toward a B1 phenotype, which instead facilitates tumour regression in the lung of tumour-bearing mice [40]. In the latter study, the activation of TLR9 in B cells caused them to become good APCs and as well as immunoglobulin-producing cells, with resultant tumour regression in a mouse model of lung carcinoma [40]. However, the modulation of B-cell-derived immunity by other TLRs in lung carcinoma needs further consideration. Understanding the role of these cells and delineating the impact of TLR-dependent signalling in the lung tumour microenvironment could open new prospective for lung cancer therapy.
Activation of TLRs on macrophages in lung cancer
Macrophages highly populate tumour microenvironment [42]. Their phenotype depends on the physiological and/or pathological condition. They can be classically active (M1) or alternatively activated (M2). In the first case, macrophages highly produce pro-inflammatory cytokines, highly express MHC molecules and have a potent tumoricidal activity [43]. Instead, the M2 phenotype is characterized by the production of high amounts of IL-10 and TGFβ, immunosuppressive cytokines, by anti-inflammatory activity and tissue repair functions [43]. Cancerous tissues are highly populated by M2 [42], which promote angiogenesis and tissue remodelling [44]. Lung macrophages express higher levels of TLR2, 3, 4, and 6 than TLR7 and TLR9 [45]. In tumour microenvironment, stimulation of macrophages with TLR ligands can drive towards the M2 phenotype [46]. Activation of the TLR2 signalling pathway facilitates lung metastasis growth, probably because of its high expression on tumour-associated macrophages (TAMs) [47]. Furthermore, TLR4-activated TAMs are responsible for the increased cancerous lesions [48]. Interestingly, TLR9 activation by CpG-ODN (type B) induces a high influx of macrophages into the lung of tumour-bearing mice [22]. MyD88 knockout mice have a reduced tumour growth correlated to the decreased influx of macrophages to the tumour environment [49]. The activation of MyD88-dependent signalling pathway targets NF-κB that maintains macrophages in an M2 phenotype [49]. Thus, it seems obvious to ask whether the activation of TRIF-dependent signalling pathway could modulate macrophage phenotype towards M1. We speculate that because IL-12 production is induced by IFN type I, the production of which is strongly stimulated by TLR3 activation, this receptor could lead to the development of the M1 phenotype within the tumour.
Activation of TLRs on myeloid-derived suppressor cells (MDSCs) in lung cancer
Myeloid-derived suppressor cells (MDSCs) as well as macrophages restrain tumour immune surveillance [50]. A very recent paper proposes that the activation of TLR2 on MDSCs facilitates tumour immune evasion, thus promoting tumour growth, in lymphoma, colon carcinoma and lung adenocarcinoma models [51]. Indeed, TLR2 knockout mice develop lower lung metastasis foci than wild type mice, after Lewis Lung Carcinoma cells (LLC1) implantation [47]. Moreover, the administration of CpG-ODN, TLR9 ligand, favours the recruitment of MDSC to the lung of tumour-bearing mice [40]. MDSCs together with Tregs may be the major responsible cells for lung tumour progression. Since both cell types can be directly or indirectly influenced by TLR activation, understanding their role in lung carcinoma could be of potential therapeutic application.
Activation of TLRs on NK cells in lung cancer
NK cells, together with the cytotoxic T lymphocytes (CTLs), are the major cellular effectors against tumours. NK cells can be activated by mDCs and pDCs via both cell–cell contact and via IFN type I [52]. Tumour regression and CTLs induction can largely depend on MyD88-dependent signalling pathway that leads to the expression of MHC I, fundamental for CTLs activity [53]. In contrast, although NK cells are barely activated by MyD88-dependent pathway, their cytotoxicity can be induced by TRIF-dependent pathway [54]. Indeed, the genetic absence of TRIF facilitated tumour progression in melanoma-bearing mice that received Poly I:C, TLR3 ligand [54]. In this latter study, the activation of TLR3 on mDCs facilitated NK cell-mediated cytotoxicity in a cell–cell contact manner [54]. However, it is also to point out that TLR3 activation can lead to the apoptosis cascade [55] that can serve as a potential strategy for tumour regression as already demonstrated in a prostate cancer mouse model after Poly I:C administration [56]. Moreover, Poly I:C can also sense cytoplasmic receptors such as MDA5 and RIG1, which contribute to NK activation [57]. Interestingly, pulmonary metastasis-bearing mice were protected when NK cells were activated through in vivo administration of Poly I:C [58]. However, since Poly I:C mimics viral moieties [31], it would be very interesting to understand how some viruses, such as human papillomavirus and respiratory syncytial virus, can precede cancer development [59]. The answer to this question may underlie TLR7 and TLR9 signalling, which expression seems to be reduced by the viral particles [59], contributing to reduced innate immune response during oncogenesis. Recent studies on TLR2 agonists including lipopeptides also revealed that stimulation of TLR2 on mDC results in activation of the MyD88 pathway in mDC to drive external NK activation [61]. However, the activation of NK cells was mediated by the production of cytokines by TLR2-MyD88 response on mDCs [60]. This effect was also observed for other TLR agonists, such as imiquimod (TLR7/8 agonist), [61] CpG-ODN (TLR9 agonist) [22], LPS (TLR4 agonist) [62], which did not induce a direct activation of NK cells. Nonetheless, the activation of NK cells after the administration of TLR3 [47], TLR7/8 [63] ligands reduced lung metastases.
Activation of TLRs on mast cells in lung cancer
Mast cells are found in many tumours [64]. They are highly represented in lung carcinoma, where their presence correlates with increased intratumoral microvessel density, enhanced tumour growth and tumour invasion, and poor clinical outcome [65]. However, the localization of mast cells into the tumour, rather than in the peritumoural area, could reflect improved survival in non-small-cell lung cancer [66]. Activation of mast cell receptors, including TLRs, can release many pro-angiogenic and tumour growth stimulatory mediators, which in turn can affect both stromal and cancerous cells by enhancing tumour proliferation. In contrast, the activation of TLR2 on mast cells countered tumour growth in a mouse model of lung carcinoma in which LLC1 cells were implanted subcutaneously [67]. Although extensive studies demonstrate a pro-tumorigenic role for mast cells, Oldford et al. [67] demonstrated that the activation of TLR2 on these cells increases IL-6 anti-tumour activities and promotes NK and CD8+ cytotoxic T-cell recruitment into the tumour mass in a CCL1-dependent manner. Though, it is well documented that IL-6 can increase the pro-tumour activity of STAT-3 [22] and that TLR2 is crucial for Treg expansion [68].
Similarly to TLR2, the activation of TLR3, TLR4, TLR7 and TLR9 [69] can increase the release of IL-5 [73], IL-13 and IgE from mast cells, which are highly implicated in the airway eosinophilic inflammation such as asthma [70], Th2-like pathology. Since lung carcinoma is a Th2-based pathology, the role of mast cells in lung tumour growth still remains controversial. Moreover, the activation of TLR9 by CpG-ODN inhibits the accumulation of peribronchial mast cells in vivo which alter lung expression of Th2 cell-derived mast cell growth factors [71]. In addition, the stimulation of TLR3 on human mast cells decreases mast cell adhesion to fibronectin and vitronectin, extracellular matrix proteins, highly produced in lung cancer and thus abrogates mast cell attachment-dependent potentiation of IgE-mediated responses [72]. It is clear that the role of mast cells and specifically the role of TLRs on mast cells can contribute to lung cancer establishment/progression.
Activation of TLRs on stromal cells in lung cancer
The stromal matrix plays a fundamental role in the development and progression of lung cancer [74]. Stromal cells modulate the tumour immune microenvironment, by ‘deciding’ the immune cell phenotype, depending on the pro-inflammatory or anti-inflammatory cytokines they produce [75].
TLRs are implicated in tissue homoeostasis and tissue remodelling [76]. TLR2 and TLR4 are crucial for the epithelial cell homoeostasis during the steady state phase. Activation of the aforementioned TLRs allow the cell to avoid death and facilitate the repair and regeneration of the tissue via the activation of the phosphoinositdyl-3-kinase (PI3 K)-Akt signalling pathway, which in turn up-modulates the expression of anti-apoptotic proteins, such as Bcl-2, IAPs [54]. PI3 K-Akt, NF-κB, MAPKs, JNKs, p38 and ERKs, all of which involved in TLR signalling, are essential for the differentiation/proliferation of lung cancer cells [77] (Fig. 3). The activation of TLR2, 4 and TLR9 has been reported to induce the release of both anti-inflammatory cytokines, such as TGFβ and IL-10, and of growth factors such as VEGF and fibroblast growth factor 2 (FGF2), involved in the development of lung cancer. Thus, since TLR2, TLR4, TLR7/8 and TLR9 are highly expressed in lung carcinoma tissues, it is likely that they can modulate tumour proliferation via the up-regulation of one of the above proteins. Indeed, TLR2 can facilitate the extracellular matrix remodelling [76] and TLR4 induces EGFR-mediated signalling [75, 76], which can allow lung carcinoma to establish and progress (Fig. 3). Interestingly, the induction of the TLR9 signalling pathway is described as beneficial for tumour regression, especially in association with cetuximab (an EGFR inhibitor: monoclonal antibody) treatment [78]; however, these studies were conducted on in vitro models and contrast with the observation that human tumour epithelial lung cells are highly proliferative in the presence of CpG-ODN, the TLR9 ligand [79]. Furthermore, the activation of TLR9 can induce VEGF release and lead to enhanced lung cancer progression in mice [80]. Moreover, no studies have been conducted on TLR3 and TLR7 signalling in lung carcinoma. It has been described that TLR3 and TLR7 activation promotes tumour regression in prostate and melanoma mouse models by inducing apoptosis [54, 76], as observed in in vitro studies on a human lung cancer cell line treated with a TLR7 ligand, imiquimod [12].

The activation of TLR2 (red arrows), TLR4 (black arrows) and TLR9 (blue arrows) on stromal cells can facilitate lung tumour progression by inducing the synthesis/release of both immune-suppressive cytokines, such as TGFβ and IL-10, which facilitate tumor progression, and extracellular matrix proteins and growth factors (i.e. VEGF, EGF and FGF2). Little is known about the role of TLR7 and TLR3 in lung carcinoma
Tumour progression depends on vascular endothelium growth factor (VEGF) that facilitates angiogenesis, a physiological process that is ‘used’ by tumour cells to survive [81]. In vitro models showed a lower proliferation of HUVEC cells when CpG-ODN and bevacizumab (VEGFR inhibitor: monoclonal antibody) are co-administered [78]. VEGF has a central role at connecting the reactive lung stroma with immune cells [81], since it is produced by cancer cells themselves but also by fibroblasts and immune cells [75, 80, 81]. Indeed, CpG-ODN-induced lung carcinoma progression is also associated with VEGF release from lung fibroblasts in mice [80]. However, although VEGF is considered a potential pro-tumour factor forming new vessels that feed the tumour mass, it can also promote vascular permeability, facilitating the influx of inflammatory cells to the lung [75], which could facilitate the anti-tumour or pro-tumour activity. Noteworthy, the extravasation of fibrin, hyaluronan acid and other extracellular matrix proteins can occur and these can be ‘sensed’ as dangerous signals (DAMPs) by TLR2 and TLR4, further exacerbating the inflammatory cascade [75, 82]. Moreover, lung VEGF functionally affects local mDCs for the transition from the innate response to a Th2-like skew [83]. TLR2 [75], TLR4 [75] and TLR9 [80] activation can induce VEGF production, but little is known about other TLRs.
The activation of TLRs on endothelial cells leads to the release of pro-inflammatory cytokines, such as IL-1, IL-6, and of chemokines, such as IL-8, and of ICAM-1 which facilitate leukocyte recruitment to the lung [83]. Interestingly, endothelial cells increase the immunosuppressive potential of activated Treg by upregulation of PD-1 and stimulation of the production of high levels of IL-10 and TGF-β after TNFα and IFNγ stimulation [84], most likely facilitating the interaction between Treg and pDCs and the further recruitment of pDCs or other immunosuppressive cells to the lung. This scenario could exacerbate lung tumour progression.
Basic fibroblast growth factor (bFGF) is another important growth factor highly detected in lung cancerous lesions [80, 85]. It is produced by fibroblasts, recruited by VEGF and other factors, such as platelet-derived growth factor (PDGF), and released by cancer cells [80, 85]. Cancer-associated fibroblasts (CAFs) are associated with tumour growth and progression as a consequence of both higher release of growth factors (i.e. VEGF, bFGF and PDGF) and release of TGFβ, IL-10 and IL-6, which enrol regulatory cells [75, 80].
IL-6 is a pro-inflammatory cytokine that guides different immune responses through the activation of signal transducer and activator of transcription 3 (STAT-3), a transcription factor constitutively activated in cancer [86]. Activation of the MyD88-dependent signalling pathway can lead to the synthesis/release of IL-6 from several types of cells, such as fibroblasts and immune cells [31, 76, 80]. It acts in an autocrine or paracrine manner, and the activation of STAT-3 can promote either a Th17-mediated immune response [18] or to the activation of the immunosuppressive MDSCs [87, 88]. The induction of a Th17 bias is concomitant to the presence of TGFβ, IL-6, IL-1 and IL-21 [17, 18]; whereas the activation of MDSCs could be associated with the presence of some DAMPs, such as hsp72 (heat shock protein), vimentin, an extracellular matrix protein produced by fibroblasts, and high-mobility group box 1 (HMGB1), which are released into the tumour microenvironment [4, 88]. TLR2 ‘senses’ hsp72 [88], HMGB1 [4] and vimentin; whereas TLR4 and TLR9 recognize HMGB1 [4]. We could speculate that the recognition of one of these DAMPs establishes an inflammatory response into the tumour microenvironment so that to accelerate lung cancer progression.
Actual clinical trials for TLR ligands
William Coley firstly described LPS as a potential anti-tumour agent [4]. The administration of LPS in Phase II clinical trials for lung cancer did not have any anti-tumour effects [22]. Similarly, CpG-ODNs failed in Phase III clinical trial for lung carcinoma patients, reflecting the discrepancy between humans and mice [25, 80]. The underlying reason could be the way CpG was administered in mice compared with humans, or the differences between human and mice immune cell phenotypes. Moreover, to our knowledge, no clinical trials are actually in progress to test the anti-tumour activity of TLR3 and TLR7 ligands in lung carcinoma.
Summary
The biology of TLR signalling in lung cancer could be fundamental, since they are implicated in tumour establishment and progression. Elucidation of their role in lung tumorigenesis could drive towards new therapeutic approaches. Based on the recent findings, TLR signalling on tumour stroma can orchestrate immune-suppressive mechanisms that favour tumour immune escape, but on the other hand, it could activate the immune system to exert anti-tumour responses. However, if chronic inflammation is at the basis of tumour progression/establishment, the main question to answer is whether the activation of TLRs on immune cells but not on stroma cells, or viceversa, could represent new therapeutic approaches for lung carcinoma. Thus, further studies are needed to better elucidate the differential effects of TLRs on specific cell types involved in lung carcinoma.
Abbreviations
- TLRs:
-
Toll-like receptors
- MyD88:
-
Myeloid differentiation factor
- TRIF:
-
TIR-domain-containing adapter-inducing interferon-β
- PAMPs:
-
Pathogen-associated molecular patterns
- DAMPs:
-
Danger-associated molecular patterns
- DCs:
-
Dendritic cells
- (pDCs):
-
Plasmacytoid dendritic cells
- hsp:
-
Heat shock proteins
- Treg:
-
T regulatory cells
- IDO:
-
Indoleamine-2, 3-dyoxigenase
- HMGB1:
-
High-mobility group box 1
References
Jahrsdorfer B, Weiner GJ (2008) CpG oligodeoxynucleotides as immunotherapy in cancer. Cancer Ther 3(1):27–32
Igney FH, Krammer PH (2002) Immune escape of tumors: apoptosis resistance and tumor counterattack. J Leuk Biol 71(6):07–20
Prendergast GC (2008) Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene 27(28):3889–3900
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30(7):1073–1081
Wang RF, Peng G, Wang HY (2006) Regulatory T cells and toll-like receptors in tumor immunity. Semin Immunol 18:136–142
Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N (2007) Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J Endocrinol 193(3):323–330
Kluwe J, Mencin A, Schwabe RF (2009) Toll like receptors, Wound healing and carcinogenesis. J mol Med 87(2):125–138
Rakoff-Nahoum S, Medzhitov R (2009) Toll-like receptors and cancer. Nat Rev Cancer 9(1):57–63
Sorrentino R, de Souza PM, Sriskandan S, Duffin C, Paul-Clark MJ, Mitchell JA (2008) Pattern recognition receptors and interleukin-8 mediate effects of Gram-positive and Gram-negative bacteria on lung epithelial cell function. Br J Pharmacol 154(4):864–871
Iwamura C, Nakayama T (2008) Toll-like receptors in the respiratory system: their roles in inflammation. Curr Allergy Asthma Rep 8(1):7–13
Yu L, Chen S (2008) Toll-like receptors expressed in tumour cells: targets for therapy. Cancer Immunol Immunther 57:1271–1278
Cherfils-Vicini J, Platonova S, Gillard M, Laurans L, Validire P, Caliandro R, Magdeleinat P, Mami-Chouaib F, Dieu-Nosjean MC, Fridman WH, Damotte D, Sautès-Fridman C, Cremer I (2010) Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest 120(4):1285–1297
Koff JL, Shao MX, Ueki IF, Nadel JA (2008) Multiple TLRs activate EGFR via a signalling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol 294:L1068–L1075
Lechmann M, Zinser E, Golka A, Steinkasserer A (2002) Role of CD83 in the immunomodulation of dendritic cells. Int Arch Allergy Immunol 129(2):113–118
Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad El-B, Lehr HA, Schmitt E, Bopp T, Kallen KJ, Herz U, Schmitt S, Luft C, Hecht O, Hohlfeld JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-John S, Renz H, Neurath MF, Galle PR, Finotto S (2005) The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest 115(2):313–325
Reiner S, Sallusto F, Lanzavecchia A (2007) Division of labor with a workforce of one: challenges in specifying effector and memory T cell fate. Science 317:622–625
Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H (2009) IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 206(7):1457–1464
Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C (2009) T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31(5):787–798
Shepard HM, Brdlik CM, Schreiber H (2008) Signal integration: a framework for understanding the efficacy of therapeutics targeting the human EGFR family. J Clin Invest 118(11):3574–3581
Eisenbarth SC, Piggott DA, Bottomly K (2003) The master regulators of allergic inflammation: dendritic cells in Th2 sensitization. Curr Opin Immunol 15(6):620–626
Piggott DA, Eisenbarth SC, Xu L, Constant SL, Huleatt JW, Herrick CA, Bottomly K (2005) MyD88-dependent induction of allergic Th2 responses to intranasal antigen. J Clin Invest 115(2):459–467
Sorrentino R, Morello S, Luciano A, Crother TR, Maiolino P, Bonavita E, Arra C, Adcock IM, Arditi M, Pinto A (2010) Plasmacytoid dendritic cells alter the antitumor activity of CpG-oligodeoxynucleotides in a mouse model of lung carcinoma. J Immunol 185(8):4641–4650
Lambrecht BN, Hammad H (2010) The role of dendritic and epithelial cells as master regulators of allergic airway inflammation. Lancet 376(9743):835–843
Sorrentino R, Morello S, Pinto A (2010) Role of plasmacytoid dendritic cells in lung-associated inflammation. Recent Pat Inflamm Allergy Drug Discov 4(2):138–143
Krieg AM (2007) Development of TLR9 agonist for cancer therapy. J Clin Invest 117(5):1184–1194
Jankowska O, Krawczyk P, Wojas-Krawczyk K, Sagan D, Milanowski J, Roliński J (2008) Phenotype of dendritic cells generated in the presence of non-small cell lung cancer antigens—preliminary report. Folia Histochem Cytobiol 46(4):465–470
Sorrentino R, Gray P, Chen S, Shimada K, Crother TR, Arditi M (2010) Plasmacytoid dendritic cells prevent cigarette smoke and Chlamydophila pneumoniae-induced Th2 inflammatory responses. Am J Respir Cell Mol Biol 43(4):422–431
Xu H, Zhang GX, Ciric B, Rostami A (2008) IDO: a double-edged sword for T(H)1/T(H)2 regulation. Immunol Lett 121(1):1–6
de Heer HJ, Hammad H, Soullié T, Hijdra D, Vos N, Willart MA, Hoogsteden HC, Lambrecht BN (2004) Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med 200(1):89–98
Smit JJ, Lindell DM, Boon L, Kool M, Lambrecht BN, Lukacs NW (2008) The balance between plasmacytoid DC versus conventional DC determines pulmonary immunity to virus infections. PLoS One 3(3):e1720
Akira S, Sato S (2003) Toll-like receptors and their signaling mechanisms. Scand J Infect Dis 35(9):555–562
Smyth MJ (2005) Type I interferon and cancer immunoediting. Nature Immunol 6:646–648
Fabricius D, Neubauer M, Mandel B, Schütz C, Viardot A, Vollmer A, Jahrsdörfer B, Debatin KM (2010) Prostaglandin E2 inhibits IFN-alpha secretion and Th1 costimulation by human plasmacytoid dendritic cells via E-prostanoid 2 and E-prostanoid 4 receptor engagement. J Immunol 184(2):677–684
Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH (2007) Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2, 3-dioxygenase. J Clin Invest 117(9):2570–2582
Tokita D, Mazariegos GV, Zahorchak AF, Chien N, Abe M, Raimondi G, Thomson AW (2008) High PD-L1/CD86 ratio on plasmacytoid dendritic cells correlates with elevated T-regulatory cells in liver transplant tolerance. Transplantation 85(3):369–377
Karanikas V, Zamanakou M, Kerenidi T, Dahabreh J, Hevas A, Nakou M, Gourgoulianis KI, Germenis AE (2007) Indoleamine 2, 3-dioxygenase (IDO) expression in lung cancer. Cancer Biol Ther 6(8):1258–1262
Gregori S, Bacchetta R, Passerini L, Levings MK, Roncarolo MG (2007) Isolation, expansion, and characterization of human natural and adaptive regulatory T cells. Methods Mol Biol 380:83–105
Lee YK, Mukasa R, Hatton RD, Weaver CT (2009) Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol 21(3):274–280
Gerondakis S, Grumont RJ, Banerjee A (2007) Regulating B-cell activation and survival in response to TLR signals. Immunol Cell Biol 85:471–475
Sorrentino R, Morello S, Forte G, Montinaro A, De Vita G, Luciano A, Palma G, Arra C, Maiolino P, Adcock IM, Pinto A. (2011) B Cells contribute to the anti-tumour activity of CpG-ODN in a mouse model of metastatic lung carcinoma. Am J Respir Crit Care Med
Inoue S, Leitner WW, Golding B, Scott D (2006) Inhibitory effects of B cells on antitumour immunity. Cancer Res 66:7741–7747
Mantovani A, Sica A (2010) Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol 22(2):231–237
Glaros T, Larsen M, Li L (2009) Macrophages and fibroblasts during inflammation, tissue damage and organ injury. Front Biosci 14:3988–3993
Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457(7225):102–106
Lee CH, Wu CL, Shiau AL (2010) Toll-like receptor 4 signaling promotes tumor growth. J Immunother 33(1):73–82
Nagaraj S, Collazo M, Corzo CA, Youn JI, Ortiz M, Quiceno D, Gabrilovich DI (2009) Regulatory myeloid suppressor cells in health and disease. Cancer Res 69(19):7503–7506
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rébé C, Ghiringhelli F (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 120(2):457–471
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nature 6:392–401
Nahoum Rakoff, Medzhitov R (2008) Role of toll-like receptors in tissue repair and tumorigenesis. Biochemistry 73:555–561
Luo JL, Maeda S, Hsu LC, Yagita H, Karin M (2004) Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 6:297–305
Li X, Jiang S, Tapping RI (2010) Toll-like receptors signalling in cell proliferation and survival. Cytokine 49:1–9
Zitvogel L, Terme M, Borg C, Trinchieri G (2006) Dendritic cell-NK cell cross-talk: regulation and physiopathology. Curr Top Microbiol Immunol 298:157–174
Akazawa T, Masuda H, Saeki Y, Matsumoto M, Takeda K, Tsujimura K, Kuzushima K, Takahashi T, Azuma I, Akira S, Toyoshima K, Seya T (2004) Adjuvant-mediated tumor regression and tumor-specific cytotoxic response are impaired in MyD88-deficient mice. Cancer Res 64(2):757–764
Akazawa T, Ebihara T, Okuno M, Okuda Y, Shingai M, Tsujimura K, Takahashi T, Ikawa M, Okabe M, Inoue N, Okamoto-Tanaka M, Ishizaki H, Miyoshi J, Matsumoto M, Seya T (2007) Antitumor NK activation induced by the toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci USA 104(1):252–257
Cheng YS, Xu F. (2010) Anticancer function of polyinosinic-polycytidylic acid. Cancer Biol Ther. 10(12)
Chin AI, Miyahira AK, Covarrubias A, Teague J, Guo B, Dempsey PW, Cheng G (2010) Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res 70(7):2595–2603
McCartney S, Vermi W, Gilfillan S, Cella M, Murphy TL, Schreiber RD, Murphy KM, Colonna M (2009) Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J Exp Med 206(13):2967–2976
Lowe DB, Shearer MH, Aldrich JF, Winn RE, Jumper CA, Kennedy RC (2010) Role of the innate immune response and tumor immunity associated with simian virus 40 large tumor antigen. J Virol 84(19):10121–10130
Hirsch I, Caux C, Hasan U, Bendriss-Vermare N, Olive D (2010) Impaired toll-like receptor 7 and 9 signaling: from chronic viral infections to cancer. Trends Immunol 31(10):391–397
Sawahata R, Shime H, Yamazaki S, Inoue N, Akazawa T, Fujimoto Y, Fukase K, Matsumoto M, Seya T (2011) Failure of mycoplasma lipoprotein MALP-2 to induce NK cell activation through dendritic cell TLR2. Microbes Infect 13(4):350–358
Ma F, Zhang J, Zhang J, Zhang C (2010) The TLR7 agonists imiquimod and gardiquimod improve DC-based immunotherapy for melanoma in mice. Cell Mol Immunol 7(5):381–388
Zanoni I, Foti M, Ricciardi-Castagnoli P, Granucci F (2005) TLR-dependent activation stimuli associated with Th1 responses confer NK cell stimulatory capacity to mouse dendritic cells. J Immunol 175(1):286–292
Hamm S, Rath S, Michel S, Baumgartner R (2009) Cancer immunotherapeutic potential of novel small molecule TLR7 and TLR8 agonists. J Immunotoxicol 6(4):257–265
Groot Kormelink T, Abudukelimu A, Redegeld FA (2009) Mast cells as target in cancer therapy. Curr Pharm Des 15(16):1868–1878
Tomita M, Matsuzaki Y, Onitsuka T (2000) Effect of mast cells on tumour angiogenesis in lung cancer. Am Thorac Surg 69:1686–1690
Welsh TJ, Green RH, Richardson D, Waller A, O’Byrne KJ, Bradding P (2005) Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J Clin Oncol 23:8959–8967
Oldford SA, Haidl ID, Howatt MA, Leiva CA, Johnston B, Marshall JS (2010) A critical role for mast cells and mast cell-derived IL-6 in TLR2-mediated inhibition of tumor growth. J Immunol 185(11):7067–7076
Tsai YG, Yang KD, Niu DM, Chien JW, Lin CY (2010) TLR2 agonists enhance CD8+Foxp3+ regulatory T cells and suppress Th2 immune responses during allergen immunotherapy. J Immunol 184(12):7229–7237
Matsushima H, Yamada N, Matsue H, Shimada S (2004) TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. J Immunol 173(1):531–541
Nigo YI, Yamashita M, Hirahara K, Shinnakasu R, Inami M, Kimura M, Hasegawa A, Kohno Y, Nakayama T (2006) Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proc Natl Acad Sci USA 103(7):2286–2291
Ikeda RK, Miller M, Nayar J, Walker L, Cho JY, McElwain K, McElwain S, Raz E, Broide DH (2003) Accumulation of peribronchial mast cells in a mouse model of ovalbumin allergen induced chronic airway inflammation: modulation by immunostimulatory DNA sequences. J Immunol 171(9):4860–4867
Kulka M, Metcalfe DD. (2006) TLR3 activation inhibits human mast cell attachment to fibronectin and vitronectin. Mol Immunol. 1579-1586
Iwamura C, Nakayama T (2008) Toll-like receptors in the respiratory system: their roles in inflammation. Curr Allergy Asthma Rep 8(1):7–13
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nature 6:392–401
Damiano V, Caputo R, Garofalo S, Bianco R, Rosa R, Merola G, Gelardi T, Racioppi L, Fontanini G, De Placido S, Kandimalla ER, Agrawal S, Ciardiello F, Tortora G (2007) TLR9 agonist acts by different mechanisms synergizing with bevacizumab in sensitive and cetuximab-resistant colon cancer xenografts. Proc Natl Acad Sci USA 104(30):12468–12473
Droemann D, Albrecht D, Gerdes J, Ulmer AJ, Branscheid D, Vollmer E, Dalhoff K, Zabel P, Goldmann T (2005) Human lung cancer cells express functionally active Toll-like receptor 9. Respir Res 6:1
Wenzel J, Uerlich M, Haller O, Bieber T, Tueting T (2005) Enhanced type I interferon signaling and recruitment of chemokine receptor CXCR3-expressing lymphocytes into the skin following treatment with the TLR7-agonist imiquimod. J Cutan Pathol 32(4):257–262
Johnson B, Osada T, Clay T, Lyerly H, Morse M (2009) Physiology and therapeutics of vascular endothelial growth factor in tumor immunosuppression. Curr Mol Med 9(6):702–707
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246:1306–1309
Sorrentino R, Morello S, Giordano MG, Arra C, Maiolino P, Adcock IM, Pinto A (2011) CpG-ODN increases the release of VEGF in a mouse model of lung carcinoma. Int J Cancer 128(12):2815–2822
Chapoval SP, Lee CG, Tang C, Keegan AD, Cohn L, Bottomly K, Elias JA (2009) Lung vascular endothelial growth factor expression induces local myeloid dendritic cell activation. Clin Immunol 132(3):371–384
Elenbaas B, Weinberg RA (2001) Heterotypic signalling between epithelial tumour cells and fibroblasts in carcinoma formation. Exp Cell Res 264(1):169–184
Li J, Ma Z, Tang ZL, Stevens T, Pitt B, Li S (2004) CpG DNA-mediated immune response in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 287(3):L552–L558
Bedke T, Pretsch L, Karakhanova S, Enk AH, Mahnke K (2010) Endothelial cells augment the suppressive function of CD4+ CD25+ Foxp3+ regulatory T cells: involvement of programmed death-1 and IL-10. J Immunol 184(10):5562–5570
Engelhardt R, Otto F, Mackensen A, Mertelsmann R, Galanos C (1995) Endotoxin (Salmonella abortus equi) in cancer patients Clinical and immunological findings. Prog Clin Biol Res 392:253–261
Liu C, Lou Y, Lizée G, Qin H, Liu S, Rabinovich B, Kim GJ, Wang YH, Ye Y, Sikora AG, Overwijk WW, Liu YJ, Wang G, Hwu P (2008) Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J Clin Invest 118(3):1165–1175
Luo JL, Maeda S, Hsu LC, Yagita H, Karin M (2004) Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 6:297–305
Li X, Jiang S, Tapping RI (2010) Toll-like receptors signalling in cell proliferation and survival. Cytokine 49:1–9
Acknowledgments
We would like to thank Dr. Giovanni Forte and Dr.Pearl Gray, who critically read our manuscript. We are thankful to the University of Salerno for providing the financial support (FARB) for current research. Dr. Rosalinda Sorrentino is supported by the University of Salerno Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pinto, A., Morello, S. & Sorrentino, R. Lung cancer and Toll-like receptors. Cancer Immunol Immunother 60, 1211–1220 (2011). https://doi.org/10.1007/s00262-011-1057-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-011-1057-8