
Abstract
CD4+ Th17 cells induce antitumor immunity leading to the eradication of established tumors. However, the mechanism of antitumour immunity and CTL activation by Th17 cells and the distinct role of Th17 and Th17-activated CTLs in antitumor immunity are still elusive. In this study, we generated ovalbumin (OVA)-specific Th17 cells by cultivating OVA-pulsed dendritic cells with CD4+ T cells derived from transgenic OTII mice in the presence of IL-6, IL-23, TGF-β, and anti-IFN-γ antibody. We demonstrated that Th17 cells acquired major histocompatibility complex/peptide (pMHC)-I and expressed RORγt, IL-17, and IL-2. Th17 cells did not have any direct in vitro tumor cell–killing activity. However, Th17 cells were able to stimulate CD8+ CTL responses via IL-2 and pMHC I, but not IL-17 signaling, which play a major role in Th17-induced preventive immunity against OVA-expressing B16 melanoma. Th17 cells stimulated the expression of CCL2 and CCL20 in lung tumor microenvironments promoting the recruitment of various inflammatory leukocytes (DCs, CD4+, and CD8+ T cells) stimulating more pronounced therapeutic immunity for early-stage (5-day lung metastases or 3 mm, s.c.) tumor than for well-established (6 mm, s.c.) tumor. The therapeutic effect of Th17 cells is associated with IL-17 and is mediated by Th17-stimulated CD8+ CTLs and other inflammatory leukocytes recruited into B16 melanoma via Th17-stimulated CCL20 chemoattraction. Taken together, our data elucidate a distinct role of Th17 and Th17-stimulated CD8+ CTLs in the induction of preventive and therapeutic antitumor immunity, which may greatly impact the development of Th17-based cancer immunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Effector CD4+ T cells are classically divided into two lineages based on distinct cytokine secretion profiles: the IFN-γ-producing Th1 lineage and IL-4/IL-13-producing Th2 lineage. Recently, a lineage of effector Th17 cells that produce IL-17A and IL-17F and express the transcription factor RORγt through activation of STAT3 by IL-6 and IL-23 have been identified [1]. IL-17 cytokines are strongly proinflammatory and induce the expression of several chemokines such as CCL2, CCL7, and CCL20. Transgenic overexpression of IL-17A in the lungs provokes proinflammatory gene expression and tissue infiltration of leukocytes [2]. In contrast, inhibition of IL-17A expression impairs host defense against bacterial infection [3] and resistance to autoimmune diseases [4, 5].
Th17 cells and IL-17 expression have been found in various human tumors [6–10]. However, the involvement of IL-17 and Th17 cells in antitumor immunity is still controversial. For example, transgenic IL-17 expression either induced tumor regression through enhanced antitumor immunity in immune-competent mice [11, 12] or promoted tumor progression through an increase in inflammatory angiogenesis in immune-deficient mice [13]. It has been demonstrated that Th17 cells were indirectly linked to antitumor immunity [14]. Tumor-specific Th17 polarized cells were found to inhibit the growth of well-established melanoma via IFN-γ production [15]. However, the nature of Th17 cell’s role in the context of antitumor immunity still remains largely unknown. Th17 cells have been shown to stimulate antitumor immunity in both prevention and therapeutic models by recruiting DCs, granulocytes, and CD4+ and CD8+ T cells [16]. However, (i) the molecular mechanism of CD8+ CTL activation by Th17 cells, (ii) the precise role of Th17 secreted IL-17 and different types of recruited leukocytes in Th17-induced antitumor immunity, and (iii) the potentially distinct role of Th17 and Th17-stimulated CD8+ CTLs in Th17-induced antitumor immunity are still unknown.
Intercellular membrane transfer through trogocytosis plays an important role in immune modulation [17]. We have recently demonstrated that CD4+ T cells derived from ovalbumin (OVA)-specific T-cell receptor (TCR) transgenic OTII mice, which were activated in vitro with OVA-pulsed DCs (DCOVA), differentiated into Th1 cells [17]. These Th1 cells acquired peptide major histocompatibility complexes I (pMHC I) and CD80 from DCOVA by DCova activation and became capable of directly stimulating CD8+ CTL responses via endogenous IL-2 and acquired pMHC I and CD80 signaling [18, 19]. However, whether Th17 cells with distinctive phenotype from Th1 cell have a similar stimulatory effect as Th1 cells on the stimulation of CD8+ CTL responses is elusive.
In this study, we generated RORγt-expressing and IL-17-secreting OVA-specific Th17 cells by cultivation of OTII CD4+ T cells with OVA-pulsed DCOVA in the presence of IL-6, IL-23, TGF-β, and anti-IFN-γ antibody. We then immunized C57BL/6 mice with these Th17 cells to assess the potential stimulatory effect on CD8+ T-cell responses and antitumor immunity in preventive and therapeutic models against OVA-expressing B16 melanoma (BL6-10OVA).
Results
Th17 acquires pMHC I complexes by DCOVA activation
Transgenic mouse OT II CD4+ T cells activated with irradiated DCOVA in the presence of IL-23/IL-6/TGF-β and anti-IFN-γ antibody expressed cell surface FasL, intranuclear RORγt, and intracellular perforin and IL-17 by flow cytometric and RT–PCR analysis (Fig. 1a, c). By using double staining for IL-17 and IFN-γ, polarized Th17 and Th1 cells expressed intracellular IL-17 and IFN-γ, respectively (Fig. 1b). These Th17 cells also secreted IL-2 (2.8 ng/ml), IL-6 (4.5 ng/ml), IL-17 (1.8 ng/ml), and TGF-β (0.2 ng/ml) by ELISA analysis, indicating that they are Th17 cells, which is consistent with a recent report showing that Th17 simultaneously expressed both IL-17 and IL-2 [20]. There was no CD11c+ DCOVA contamination in these purified Th17 population (Fig. 1d). In addition, these Th17 cells did display pMHC I (Fig. 1a), indicating that they may acquire pMHC I from DCOVA upon DCOVA activation. This was confirmed by evidence that CD4+ T cells derived from pMHC I-negative (Kb−/−)DCOVA activation did not express pMHC I (Fig. 1e).

Phenotypic analysis of OVA-specific Th17 cells. a Naïve CD4+ T cells and in vitro DCOVA-activated Th17 were stained with a panel of antibodies (solid lines) and analyzed by flow cytometry. Irrelevant isotype-matched biotin-conjugated antibodies were used as controls (light dotted lines). b In vitro DCOVA-activated Th1 and Th17 cells were double stained with FITC-anti-IL-17 Ab and PE-anti-IFN-γ Ab and analyzed by flow cytometry. c RNA extracted from DCOVA-activated Th17 and ConA-stimulated CD4+ T (control) cells were analyzed by RT–PCR to assess the expression of ROR-γt. d DCOVA and Th17 were stained with PE-antiCD4 and FITC-anti-CD11c Abs and analyzed by flow cytometry. e Th17, (Kb−/−), Th17 cells, and CD3/CD28 bead-activated Th17 (bead-Th17) cells were stained with FITC-anti-pMHC I antibody (solid lines), and irrelevant isotype-matched antibody was used as control (dotted lines). One representative experiment of two experiments is shown
Th17 stimulates in vitro CD8+ T-cell proliferation via IL-2 and pMHC I, but not IL-17 signaling
We previously demonstrated that DCova-activated Th1 with acquired pMHC I stimulated CTL responses via IL-2 and pMHC I signaling [18, 19]. To assess Th17’s stimulatory effect, we performed 3H-thymidine incorporation assay. DCova-activated Th17 with acquired pMHC I also stimulated in vitro OTI CD8+ T-cell proliferation in a dose-dependent fashion. Interestingly, (Kb−/−)Th17 without acquired pMHC I or Th17 in the presence of anti-IL-2 Ab, but not anti-IL-17 Ab, failed to stimulate CD8+ T-cell proliferation (Fig. 2a), indicating that the in vitro Th17’s stimulatory effect on CTLs is via IL-2 and pMHC I, but not via IL-17 signaling.

Functional effect analysis of OVA-specific Th17 cells. a Irradiated DCOVA, Th17, (IL-2−/−)Th17 and (Kb−/−)Th17, and their 2-fold dilutions were co-cultured with OTI CD8+ T cells. After 2 days, the proliferative responses of CD8+ T cells were determined by overnight 3H-thymidine uptake assay. b Th17-activated CD8+ T cells with or without preincubation of concanamycin A (CMA, 1 μM) or emetin (5 μM) for 2 h were used as effector (E) cells, while 51Cr-labeled EG7 and EL4 cells were used as target (T) cells. c DCOVA-activated Th17 and Th1 were used as effector (E) cells, while 51Cr-labeled OVAII-pulsed LB27 cells and LB27 cells were used as target (T) cells. *P < 0.01 and **P > 0.05 versus cohorts of ‘no inhibitor’ group (Student’s t test). d In tetramer staining assay, the tail blood samples of wild-type B6 or DT-treated DTR-CD11c mice (6 per group) transferred with Th17 were stained with PE-H-2Kb/OVA I (PE-tetramer) and FITC-anti-CD8 Ab (FITC-CD8) and then analyzed by flow cytometry. e In tetramer staining assay, the tail blood samples from wild-type B6 mice (6 per group) transferred with DCova-activated Th17, Th17 with various KO and CD3/CD28 bead-activated Th17 (bead-Th17) or from anti-IL-17 Ab-treated B6 mice transferred with Th17 or perforin−/− mice were stained with PE-H-2Kb/OVA I (PE-tetramer) and FITC-anti-CD8 Ab (FITC-CD8) and then analyzed by flow cytometry. The value represents the percentage of tetramer-positive CD8+ T cells in the total CD8+ T-cell population with standard deviation in parenthesis. In in vivo cytotoxicity assay (in both panel d and e), the residual CFSEhigh (H) and CFSElow (L) target cells remaining in the recipients’ spleens (6 per group) were analyzed by flow cytometry. The value in each panel represents the percentage of CFSEhigh vs CFSElow target cells remaining in the spleen. (n = 6, average ± SD). *P < 0.05 versus cohorts of mice immunized with Th17 (Student’s t test). One representative experiment of two is shown
Th17-activated CD8+ T, but not Th17, cells have in vitro cytotoxicity
Since Th17 expressed cytotoxic FasL and perforin, they may have killing activity to pMHC II-expressing target cells. To assess their killing effect, we performed a chromium release assay. We found that Th17-activated CTLs showed killing activity to OVA-expressing EG7 tumor cells, and the killing activity was significantly (P < 0.01) or slightly (P > 0.05) reduced when T cells were pre-incubated with CMA or emetin, indicating that CD8+ T-cell-mediated killing activity is mainly via perforin pathway (Fig. 2b) [21]. In addition, we found that Th1 [19], but not Th17, killed OVAII peptide-pulsed LB27 cells (Fig. 2c), indicating that Th17 do not have any direct killing activity to tumor cells.
Th17 stimulates the host DC-independent CD8+ CTL responses
To assess DCova-activated Th17’s ability to induce in vivo CD8+ T-cell proliferation, we i.v. transferred B6 mice with DCova and Th17 and then performed an OVA-specific tetramer staining assay to detect OVA-specific CD8+ T-cell proliferation [19]. As shown in Fig. 2d, DCova and Th17 stimulated OVA-specific CD8+ T cells accounted for 1.43 and 0.98% of the total CD8+ T-cell population, respectively. To assess whether the host DCs are involved in Th17-stimulated CTL responses by the uptake of antigen epitopes of Th17, we i.v. transferred the transgenic DTR-CD11c mice with DT treatment for complete depletion of endogenous DCs and macrophages (Clin Exp Immunol 141: 398, 2005) with Th17 and then performed an OVA-specific tetramer staining assay to detect OVA-specific CD8+ T-cell proliferation. We found that Th17-stimulated OVA-specific CTL responses in PBS- and DT-treated DTR-CD11c mice with and without endogenous APCs were similar (Fig. 2d), indicating that Th17 stimulates the host DC-independent CD8+ CTL responses.
Th17 stimulates CTL-mediated preventive antitumor immunity via IL-2 and pMHC I, but not via IL-17 signaling
To elucidate the molecular mechanism of Th17-stimulated CTL responses, DCova-activated Th17, (IL-2−/−)Th17 with IL-2 deficiency, (Kb−/−)Th17 without acquired pMHC I, and CD3/CD28 bead-activated Th17 (bead-Th17) without pMHC I expression (Fig. 1e) were used in the in vivo proliferation and cytotoxicity experiments. To assess the involvement of IL-17, Th17 cell transferred mice were treated with anti-IL-17 Ab to block IL-17 effect. As shown in Fig. 2e, the stimulatory effect was significantly reduced in mice transferred with (Kb−/−)Th17, bead-Th17, and (IL-2−/−)Th17 (P < 0.05), but not in Th17-transferred mice with the treatment of anti-IL-17 Ab, indicating that endogenous IL-2 and acquired pMHC I, but not IL-17 signaling influence in vivo Th17’s stimulatory effect. In in vivo cytotoxicity assay, we found that Th17-transferred mice showed substantial loss of OVA-specific CFSEhigh cells, indicating that Th17 can stimulate CD8+ T-cell differentiation into effector CTLs with killing activity for OVA-specific target cells in vivo. To assess the pathway responsible for the killing activity of CD4+ Th17-stimulated CD8+ T cells in vivo, we also transferred CD4+ Th17 cells into perforin−/− mice and repeated the above-stated tetramer staining and in vivo cytotoxicity assays. We found that OVA-specific CD8+ T-cell in vivo proliferation in C57BL/6 and perforin−/− mice were similar (Fig. 2e). However, CD8+ T-cell-induced killing activity to OVA-specific CFSEhigh target cells was lost in perforin−/− mice (Fig. 2e), indicating that the in vivo CD4+ Th17-stimulated CD8+ T-cell-induced killing activity to OVA-specific target cells is via perforin-dependent pathway. Interestingly, (IL-2−/−)Th17-, (Kb−/−)Th17-, and bead-Th17-transferred mice maintained their OVA-specific CFSEhigh target cell numbers, indicating that in vivo stimulatory effect of Th17 is mediated by its IL-2 secretion and pMHC I targeting. To assess preventive antitumor immunity, we performed animal studies by i.v. injection of BL6-10OVA cells into the above-transferred mice 6 days subsequent to transfer. We found that all mice (8/8) were free from metastasis, whereas all (8/8) Th17-transferred mice with treatment of anti-CD8 Ab completely lost their antitumor immunity (Exp I of Table 1), indicating that Th17-induced antitumor immunity is mainly mediated by CD8+ T cells. We also found that all (8/8) (IL-2−/−)Th17- and (Kb−/−)Th17-transferred, but not mice with treatment of anti-IL-17 Ab, lost their antitumor immunity (Exp I of Table 1), indicating that Th17’s stimulatory effect on preventive antitumor immunity is also mediated by IL-2 (not IL-17) signaling and pMHC I targeting. To assess the long-term immunity, we also challenged Th17-transferred mice 60 days after the primary immunization. As shown in Exp II of Table 1, all transferred mice had a long-term protective antitumor immunity.
IL-17 is associated with DCova-activated Th17-induced eradication of early-stage (5 day) lung tumor metastases
To assess Th17’s therapeutic effect, we i.v. injected C57BL/6 mice with BL6-10OVA tumor cells. Five days after tumor cell injection, mice were i.v. transferred with DCova-activated Th17 cells with pMHC I expression and CD3/CD28 bead-activated Th17 (bead-Th17) cells without pMHC I expression. Lung tumor colonies were numerated 10 days after transfer. Compared with untreated control mice, those mice transferred with DCova-activated Th17, but not with bead-Th17, had significantly fewer tumor foci (P < 0.05) (Fig. 3a), indicating that DCova-activated Th17 cells have efficient therapeutic effect on early-stage (5 day) tumor lung metastasis via acquired pMHC I signaling. To assess the role of Th17-secreted IL-17 in DCova-activated Th17-induced antitumor immunity, we treated immunized mice with anti-IL-17 Ab to block IL-17 signaling. We found that Th17-transferred mice with treatment of anti-IL-17 Ab had numerous tumor foci as the control mouse group (Fig. 3a), indicating that IL-17 is critically involved in DCova-activated Th17-induced therapeutic antitumor immunity.

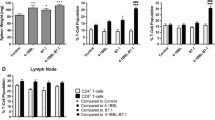
Therapeutic effect of OVA-specific Th17 cells on eradication of established lung tumor metastasis. a C57BL/6 mice or Iab−/− KO mice or toxin-treated DTR-CD11c mice or perforin−/− mice bearing 5-day lung B16 melanoma were i.v. immunized with DCova-activated Th17 with or without various Abs against CD8, CD11c, IL-17 for depletion of host CD8+ T cells, DCs, and cytokine, respectively. C57BL/6 mice bearing 5-day lung B16 melanoma were also i.v. immunized with CD3/CD28 bead-activated Th17 (bead-Th17). Tumor colonies were counted on day 16 after tumor challenge (n = 8, average ± SD). *P < 0.05, versus cohorts of tumor foci in mice injected with control Ab (Student’s t test). b mRNA gene expression analysis was assessed by RT–PCR. Data shown were normalized to the reference gene GAPDH. Graphs represent the average values of four mice after duplicate analysis per sample (n = 4, average ± SD). c Total numbers of leukocytes from cell fractions of tumor-bearing lungs were calculated from percentages of live cells gated on CD45.2 (n = 4, average ± SD). d 5-day lung tumor-bearing mice were transferred with 3 × 106 OTI CD8+ T cells and after 3 days percentages of OTI CD8+ T cells out of total CD8+ T-cell population in lungs were measured by gating on FITC-CD8 and PE-tetramer-positive T cells (n = 4, average ± SD). One representative experiment of two is shown
Th17 induces recruitment of leukocytes into tumors via Th17-stimulated CCL2/CCL20 expression
To assess the potential Th17 stimulated CCL2/CCL20 expression, we first analyzed the expression of CCL2/CCL20 by lung cells using real-time PCR. We found that BL6-10 tumor cells and normal lungs did not express any CCL2/CCL20 chemokines (Fig. 3b). However, the expression of CCL2/CCL20 was greatly increased in lung cell fractionations containing both tumor and lung cells, indicating that Th17 cells stimulate expression of CCL2/CCL20. Further analysis of lung leukocyte fractionations by flow cytometry revealed that CD11c+ DCs, CD4+, and CD8+ T cells were significantly increased in Th17-treated mouse lungs (Fig. 3c) (P < 0.05). To assess the potential recruitment of OVA-specific CD8+ T cells, we transferred DCOVA-activated OTI CD8+ T cells into B6 mice bearing 5-day lung tumor metastasis and then numerated tumor-infiltrating CD8+ T cells by flow cytometry. We found that Th17 significantly promoted tumor infiltration of OVA-specific CD8+ T cells indicating that Th17 cells promotes tumor infiltration of OVA-specific CD8+ T cells via CCL20 chemoattraction.
Th17-activated CD8+ T cells mediate therapeutic effect via perforin-dependent pathway on early-stage (5 day) lung tumor metastases
To assess the role of recruited DCs, CD4+, and CD8+ T cells, the above immunized C57BL/6 mice were treated with different antibodies to deplete CD8+ T cells or DCs. Alternatively, DTR-CD11c transgenic mice with the treatment of toxin to deplete the host DCs were transferred with Th17. To assess the role of recruited CD4+ T cells in Th17-induced therapeutic effect, we also i.v. transferred Iab−/− KO mice with Th17, which lack the host CD4+ T cells. We found that only the mice treated with anti CD8 Ab (host CD8+ T cells depleted), but not mice with depletion of host DCs (anti CD11c Ab or DTR-CD11c mice treated with toxin) or lacking host CD4+ T cells (Iab−/− KO mice), showed numerous tumor foci (Fig. 3a), indicating that Th17-stimulated CD8+ CTLs play an important role, but the host DCs and CD4+ T cells recruited into tumor tissues are not involved in Th17-induced therapeutic effect. To assess the molecular mechanism of CD8+ T-mediated killing, we repeated Th17 treatment in tumor-bearing perforin−/− mice, where Th17-stimulated host OVA-specific CD8+ T cells are perforin deficient. We found that Th17-transferred perforin−/− mice showed numerous tumor foci (Fig. 3a), indicating that Th17-stimulated CTLs mediate therapeutic effect via perforin-dependent pathway.
Th17-activated CD8+ T cells are potent in the eradication of early-stage (3 mm), but not well-established (6 mm), s.c. tumors
To determine whether Th17 protects against tumors in different tissues in addition to lung tissues, we s.c. inoculated B6 mice with BL6-10OVA cells. We then treated mice bearing different sizes (early stage: 3 mm and well-established tumor: 6 mm) of B16 melanoma with Th17. We found that all tumor (3 mm)-bearing mice (8/8) died of tumor within 21 days without treatment, whereas all 8/8 tumor-bearing mice with treated with Th17 survived (Fig. 4a), indicating that DCova-activated Th17 cells have immunotherapeutic effect on early-stage tumors. To assess the role of IFN-γ expression, and host CD4+ and CD8+ T cells in the immunotherapeutic effect, we used IFN-γ−/−, Iab−/−, and H-2 Kb−/− KO mice in the above experiments. We found that DCova-activated Th17 cell-induced therapeutic effect was not affected in IFN-γ−/− and Iab−/− KO mice (Fig. 4b), indicating that Th17-induced therapeutic effect is not mediated via host IFN-γ and CD4+ T cells. However, its therapeutic effect was completely lost in H-2 Kb−/− KO mice lacking host CD8+ T cells (Fig. 4b), confirming that Th17-stimulated host CD8+ CTLs play a major role in Th17-induced therapeutic antitumor immunity. To assess the potential immunotherapeutic effect on well-established tumors, we repeated experiments in mice bearing BL6-10OVA tumors (6 mm). We found that none (0/8) of the treated mice survived though their survival was significantly prolonged (P < 0.05) (Fig. 4c), indicating that DCova-activated Th17 cells, though having therapeutic effect, are not potent enough in well-established tumors.

Therapeutic effect of OVA-specific Th17 cells on the eradication of established s.c. tumors. a Wild-type C57BL/6 mice (n = 8) bearing early-stage (3 mm) B16 melanoma were i.v. immunized with DCova-activated Th17 cells or PBS as a control. b Wild-type C57BL/6, IFN-γ−/−, H-2 Kb−/−, and Iab−/− gene KO mice (n = 8) bearing early-stage (3 mm) B16 melanoma were i.v. immunized with DCoav-activated Th17 cells. Tumor regression or growth was monitored. The evolution of the tumors in individual mouse is depicted for their survival period. c Wild-type C57BL/6 mice (n = 8) bearing well-established (6 mm) B16 melanoma were i.v. immunized with Th17. Tumor size was measured daily using an engineering caliper. The evolution of the tumors in individual mouse is depicted for their survival period. *P < 0.05 versus cohorts of control group (Log rank test). One representative experiment of three is shown
Discussion
Th17 cells are an important inflammatory component and have been shown to promote inflammation in autoimmune diseases [22, 23]. Recent evidence suggests that Th17 cells are also involved in tumor immunology and may be a target for cancer immunotherapy [24]. We have previously demonstrated that Th1 cells acquired pMHC I complexes and co-stimulatory molecules from DCOVA upon DCOVA activation and became capable of stimulating OVA-specific CD8+ CTL responses via IL-2 and pMHC I signaling and antitumor immunity [15, 19]. In our current study, we demonstrated for the first time that (i) in vitro DCOVA-activated Th17 cells expressing RORγt, IL-17, and IL-2 also acquire pMHC I complexes upon activation by DCOVA and (ii) Th17 with cytokine profile distinct from Th1 is also capable of stimulating CD8+ CTL responses and long-term memory via IL-2 and pMHC I, but not IL-17 signaling. Endogenous IL-2 of Th17 cells is important in CTL induction, though Il-2 has been shown to constrain Th17 generation from CD4+ T precursors via STAT5 [25]. In addition, we also demonstrated that Th17-induced preventive antitumor immunity is mainly mediated by Th17-stimulated CTLs.
In the therapeutic model for early-stage (5 day) lung tumor metastases, we found that (i) it is Th17-activated CD8+ T cells that play a major role in the eradication of lung metastatic tumors and (ii) Th17 stimulated the expression of CCL2/20 in lung tumor microenvironments, which promoted the recruitment of various inflammatory leukocytes (DCs, CD4+, and CD8+ T cells) to induce therapeutic immunity. Our study elucidates the molecular mechanism of Th17’s stimulatory effect on CD8+ CTL responses and also demonstrated that (i) it is the Th17-stimulated CTLs, but not Th17 cells, that themselves have direct in vitro killing activity to tumor cells, (ii) Th17-secreted IL-17, but not the host IFN-γ, is associated with Th17-induced therapeutic effect, and (iii) although Th17 cells promote tumor infiltration of various inflammatory leukocytes, the tumor-specific CD8+ T cells with killing activity via perforin pathway [26], but not DCs and CD4+ T cells recruited via CCL20 chemoattraction, play a major role in Th17-induced therapeutic effect. To date, adoptive T-cell immunotherapy for cancer by using in vitro expanded tumor-infiltrating CD8+ T cells has achieved some degree of success [27, 28]. However, one of the major obstacles in this therapy is only very limited number of transferred CD8+ T cells that eventually infiltrate into tumors [29, 30], which greatly affects its therapeutic efficacy. Therefore, Th17 cells may be useful in enhancing adoptive CD8+ T-cell immunotherapy for cancer.
Th17-polarized cells derived from TRP-1-specific TCR transgenic mice inhibited the growth of large s.c. established B16 melanoma (~0.6 cm2, equal to ~7–8 mm in diameter) after adoptive transfer of Th17 cells [15]. However, in our current study, we found that Th17 cells efficiently cured s.c B16 melanoma only in early stage (3 mm), but not well-established stage (6 mm), which is consistent with a previous report on the eradication of 6-day s.c. tumors [31]. The discrepancy in therapeutic efficiency may be due to the different treatment protocols in these reports. In our protocol, we simply i.v. transferred OVA-specific Th17 into B16 melanoma-bearing mice for direct assessment of Th17 therapeutic effect. In their protocol [15], they have combined adoptive transfer of Th17 with an extra-total body sublethal irradiation plus TRP-1 virus and IL-2 administration. Their complex protocol will definitely interfere with the assessment of Th17-mediated therapeutic effect. For example, sublethal irradiation will induce lymphopenia leading to proliferation and prolonged survival of transferred T cells [32, 33] and conversion of Th17 into Th1 [34, 35], whereas TRP-1 virus vaccine alone can activate both Th1 and CTL responses important for tumor rejection [36, 37].
Based upon previous reports and our own findings, we propose the following model for Th17-induced antitumor immunity: (i) Th17 directly stimulates tumor-specific CD8+ T-cell responses via pMHC I and IL-2 signaling, (ii) homing molecule (CXCR4, CCR6, and CD161)-expressing Th17 migrates into tumors [20, 38] by tumor environmental RANTES and MCP-1 chemoattraction [39], (iii) tumor-infiltrating Th17 stimulates tumor tissues to express CCL20 for recruiting CCR6-expressing tumor-specific CD8+ T cells into tumors via CCL20 chemoattraction, and (iv) tumor-specific CD8+ T cells exert direct killing activity to tumor cells via perforin/granzyme B pathway (Fig. 5).

Mechanism of Th17-mediated antitumor immune responses. Tumor-infiltrating Th17 cells stimulate tumor microenvironment to express CCL2/20 leading to the recruitment of inflammatory cells such as CD8+ CTLs derived from direct Th17 stimulation via pMHC I and IL-2 signaling into the tumor site, which exert direct killing activity to tumor cells via perforin/granzyme B pathway
Taken together, our data demonstrate a distinct role of Th17 and Th17-stimulated CD8+ T-cell responses in preventive and therapeutic antitumor immunity, which may greatly impact the development of Th17-based cancer immunotherapy.
Materials and methods
Reagents, antibodies, cell lines, and animals
The biotin-labeled anti-CD4 (GK1.5), CD11c (HL3), CD25 (7D4), CD40L (TRAP1), CD69 (H1.2F3), FasL (CD178), and major histocompatibility complex (MHC) class I (H-2 Kb) (AF6-88) antibodies (Abs) were purchased from BD Biosciences (San Diego, CA). The R-phycoerythrin (PE)-conjugated anti-mouse IFN-γ (XMG1.2), perforin (δG9), and FITC-conjugated IL-17 (TC11-18H10) Abs were obtained from Pharmingen Inc. (Mississauga, Ontario, Canada). The PE-conjugated anti-mouse RORγt (RORg2) Abs were purchased from BioLegend (San Diego, CA). The FITC-conjugated avidin was obtained from Jackson Immuno Research Laboratory Inc. (West Grove, PN). Peptides OVAI (OVA257–264) specific for H-2Kb, OVAII (OVA323–339) specific for Iab, and 3LL lung carcinoma antigen (Ag) Mut1 peptide specific for H-2Kb were synthesized by Multiple Peptide Systems (San Diego, CA). The FITC-labeled anti-CD8 Ab and PE-H-2Kb/OVAI tetramer were obtained from Beckman Coulter (Mississauga, Ontario, Canada). Recombinant cytokines were obtained from R&D Systems Inc. (Minneapolis, MN). Tumor cell lines including ovalbumin (OVA)-expressing thymoma (EG7) and BL6-10OVA and Iab-expressing LB27 were available in our laboratory [40]. Wild-type C57BL/6, IL-2+/−, perforin−/−, Iab−/− and H-2Kb−/− knockout (KO), diphtheria toxin receptor (DTR)-CD11c transgenic mice [41], and OVA-specific T-cell receptor (TCR) transgenic OTI and OTII mice, which express TCR specific for OVAI and OVAII, respectively, were all purchased from the Jackson Laboratory (Bar Harbor, MA). Homozygous OTII/H-2Kb−/− and OTII/IL-2−/− mice were generated by backcrossing the designated gene KO mice onto OTII background. All mice were treated according to Animal Care Committee guidelines of University of Saskatchewan.
Preparation of dendritic cells
Bone marrow–derived dendritic cells (DCs) of C57BL/6 mice were generated in the presence of GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) as described previously [18, 19]. These DCs expressed MHC II, CD40, and CD80, indicating that they were mature DCs. They were then pulsed with OVA (0.3 mg/ml) overnight at 37°C and termed as DCOVA. OVA-pulsed DCs generated from H-2Kb gene KO mice were referred to as (Kb−/−)DCOVA.
Preparation of OVA-specific T cells
To generate OVA-specific Th17 cells, naïve CD4+ T cells (2 × 105 cells/ml) from OT II mice were stimulated for 3 days with irradiated (4,000 rads) DCOVA (1 × 105 cells/ml) in the presence of IL-6 (10 ng/ml), IL-23 (10 ng/ml), TGF-β (10 ng/ml), and anti-IFN-γ antibody (20 μg/ml). These DCova-activated Th17 cells were purified by positive selection using CD4 microbeads (Miltenyi Biotech, Auburn, CA). (Kb−/−)DCOVA-activated CD4+ T cells derived from OTII mice were termed (Kb−/−)Th17, whereas DCOVA-activated CD4+ T cells derived from OTII/IL-2−/− mice were termed (IL-2−/−)Th17. Alternatively, naïve CD4+ T cells (2 × 105 cells/ml) from OT II mice were stimulated with CD3/CD28 T-cell expander Dynabeads (Invitrogen) at a ratio of 1:1 (bead:cell) for 4–5 days in the presence of IL-6 (10 ng/ml), IL-23 (10 ng/ml), and TGF-β (10 ng/ml) [42]. The cytokine profiles of the above various gene KO Th17 or CD3/CD28-bead-activated Th17 (bead-Th17) were similar to the cytokine profile of DCova-activated Th17 cells except for the specific molecule deficiency (data not shown). Preparation of DCOVA-activated OVA-specific Th1 cells expressing IL-2, IFN-γ, FasL, and perforin, but not IL-4 and OVA-specific CD8+ T cells, were previously described [19, 43].
Phenotypic characterization of OVA-specific Th17 cells
The above Th17 cells and Th17 cells with various gene KO were stained with antibodies and analyzed by flow cytometry. To measure intracellular expression of cytokines, Th17 were processed using a intracellular staining commercial kit (BD Biosciences, San Diego, CA) and stained with anti-perforin and RORγt Ab or double stained with FITC-conjugated anti-IL-17 Ab and PE-conjugated anti-IFN-γ Ab. Culture supernatants of Th17 re-stimulated with irradiated (4,000 rads) OVAII peptide-pulsed LB27 cells [18, 19] were analyzed for cytokine expression using ELISA kits (Endogen, Cambridge, MA).
CD8+ T-cell proliferation assays
To assess the functional effect of OVA-specific Th17, we performed in vitro CD8+ T-cell proliferation assay. Twofold serially diluted irradiated (4,000 rads) DCOVA, Th17, (IL-2−/−),Th17, and (Kb−/−)Th17 cells (0.4 × 105 cells/well) were co-cultured with a constant number of naive OT I CD8+ T cells (1 × 105 cells/well). After culturing for 48 h, overnight thymidine incorporation was quantified by liquid scintillation counting. In in vivo proliferation assay, C57BL/6 or perforin−/− mice (6 per group), or transgenic DTR-CD11c mice (6 per group) with a single dose (4 ng/g mouse body weight) of i.v. diphtheria toxin (DT) treatment 1 day before Th17 transfer were i.v. transferred with DCOVA (1 × 106 cells), DCova-activated Th17 (3 × 106 cells) or Th17 (3 × 106 cells) with various gene KO or CD3/CD28 bead-activated Th17 (bead-Th17) (3 × 106 cells). Six days subsequent to transfer, tail blood samples were stained with FITC-anti-CD8 Ab and PE-H-2 Kb/OVAI tetramer and analyzed by flow cytometry.
Cytotoxicity assays
In in vitro cytotoxicity assay, DCOVA-activated OTII Th17 and Th1 were used as effector (E) cells, while 51Cr-labeled OVAII-pulsed LB27 cells were used as target (T) cells. In another experiment, Th17-activated OTI CD8+ cytotoxic (Tc) cells were used as effector (E) cells, while 51Cr-labeled EG7 were used as target (T) cells in a chromium release assay. CMA and emetin inhibitors were used in Th17-activated OTI CD8+ cytotoxic (Tc) cell cytotoxicity assay to inhibit perforin and FAS ligand–mediated cytotoxicity, respectively [19]. In in vivo cytotoxicity assay, 6 days following Th17 transfer, the Th-transferred C57BL/6 mice (6 per group) were i.v. injected with a 1:1 (OVA-specific CFSEhigh:nonspecific control CFSElow) mixture of splenocyte targets. Sixteen hours subsequent to target cell delivery, the proportion of CFSEhigh and CFSElow target cells remaining in the spleens was analyzed by flow cytometry [21].
Real-time RT–PCR
Total RNA was extracted from BL6-10OVA cells, lung cell fraction from normal or tumor-bearing lungs with Qiagen RNeasy purification kit (Qiagen, Mississauga, Ontario, Canada) as per manufacturer’s protocols. The primer sets for real-time PCR analysis of RORγt, CCL2, and CCL20 were designed as previously described [16]. Qiagen quantitative reverse transcription kit (Qiagen) was used to synthesize cDNA, which was then analyzed by real-time quantitative PCR in triplicates by using SYBR Green PCR mastermix (Applied Biosystems, Foster City, CA, USA) in the Stepone Real-time PCR system (Applied Biosystems). Each gene expression was normalized to GAPDH expression level using comparative CT method.
Lung fractionation and cell analysis
Mouse lungs were digested with collagenase D (1 mg/ml, Worthington Biochemical, Freehold, NJ) at 37°C for 30 min and 5 min with 0.01 M EDTA for prevention of DC–T-cell aggregate formation [44]. The cells were separated using Histopaque (Sigma, St. Louis, MO). The middle section of the gradient, which was enriched with leukocytes, was counted and analyzed by flow cytometry, whereas the bottom fraction that was enriched with tumor cells and lung cells was used for RNA extraction.
Animal studies
In preventive immunity model, wild-type C57BL/6 mice (8 per group) were i.v. transferred with OVA-specific Th17 (3 × 106), Th17 (3 × 106) with various gene KO and Th17 (3X106) with various antibodies. Six days subsequent to transfer, mice were i.v. challenged with BL6-10OVA cells (0.5 × 106). Mice were killed after 3 weeks, and the numbers of metastatic lung tumor colonies were counted [18]. In the lung tumor metastasis therapeutic model, C57BL/6 mice (8 per group) were i.v. injected with BL6-10OVA tumor cells (0.5 × 106). After 5 days of tumor cell injection (5-day lung tumor metastasis), mice were i.v. injected with Th17 or CD3/CD28 bead-activated Th17 (bead-Th17) (3 × 106 cells/mouse) cells. To deplete CD8+ T cells or DCs or to block IL-17, C57BL/6 mice were i.p. injected with anti-CD8 or anti-CD11c or anti-IL-17 Ab (each, 0.5 mg/mouse) 1 day before Th17 transfer and followed by another two injections (once every 3 days). To assess the involvement of host CD4+ T cells or DCs or host CD8+ T-cell’s perforin in therapeutic effect, Iab−/− KO mice (lacking CD4+ T cells) or DTR-CD11c transgenic mice (8 per group) with diphtheria toxin (DT) treatment (a single dose of 4 ng/g body weight of mouse; i.p.) to deplete host CD11c+ DCs or perforin−/− mice were used [45]. Mice were killed on day 16 after i.v. injection of tumor cells. The metastatic lung tumor colonies were counted. In the s.c. tumor therapeutic model, C57BL/6 or Kb−/− or Iab−/− or IFN-γ−/− KO mice (8 per group) were s.c. injected with BL6-10OVA tumor cells (0.5 × 106). When s.c. tumors reached 3 or 6 mm in diameter, these mice were i.v. injected with Th17 cells (3 × 106 cells). Tumor growth was monitored by measuring tumor diameter using caliper; for ethical reasons, all mice with tumors that achieved a size of 1.5 cm in diameter were killed.
Statistical analysis
Mouse survival was analyzed using log rank test [46, 47], and all other experiments were tested for statistical differences using unpaired, two-tailed, Student’s t test. Differences were considered significant if P < 0.05.
References
Weaver CT, Hatton RD, Mangan PR, Harrington LE (2007) IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 25:821–852
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK (2001) Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol 25:335–340
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C (2008) Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29:44–56
Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M (2006) IL-23 promotes tumour incidence and growth. Nature 442:461–465
Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J, Chang A, Zou W (2007) Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol 178:6730–6733
Miyahara Y, Odunsi K, Chen W, Peng G, Matsuzaki J, Wang RF (2008) Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc Natl Acad Sci USA 105:15505–15510
Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, Meeker AK, Isaacs WB, Drake CG (2008) Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res 14:3254–3261
Zhang B, Rong G, Wei H, Zhang M, Bi J, Ma L, Xue X, Wei G, Liu X, Fang G (2008) The prevalence of Th17 cells in patients with gastric cancer. Biochem Biophys Res Commun 374:533–537
Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautes-Fridman C, Fossiez F, Haicheur N, Fridman WH, Tartour E (2002) Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood 99:2114–2121
Hirahara N, Nio Y, Sasaki S, Minari Y, Takamura M, Iguchi C, Dong M, Yamasawa K, Tamura K (2001) Inoculation of human interleukin-17 gene-transfected Meth-A fibrosarcoma cells induces T cell-dependent tumor-specific immunity in mice. Oncology 61:79–89
Numasaki M, Watanabe M, Suzuki T, Takahashi H, Nakamura A, McAllister F, Hishinuma T, Goto J, Lotze MT, Kolls JK, Sasaki H (2005) IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol 175:6177–6189
Spolski R, Leonard WJ (2008) Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol 26:57–79
Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, Gattinoni L, Wrzesinski C, Hinrichs CS, Kerstann KW, Feigenbaum L, Chan CC, Restifo NP (2008) Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 112:362–373
Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C (2009) T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31:787–798
Ahmed KA, Munegowda MA, Xie Y, Xiang J (2008) Intercellular trogocytosis plays an important role in modulation of immune responses. Cell Mol Immunol 5:261–269
Xiang J, Huang H, Liu Y (2005) A new dynamic model of CD8+ T effector cell responses via CD4+ T helper-antigen-presenting cells. J Immunol 174:7497–7505
Umeshappa CS, Huang H, Xie Y, Wei Y, Mulligan SJ, Deng Y, Xiang J (2009) CD4+ Th-APC with acquired peptide/MHC class I and II complexes stimulate type 1 helper CD4+ and central memory CD8+ T cell responses. J Immunol 182:193–206
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114:1141–1149
Huang H, Bi XG, Yuan JY, Xu SL, Guo XL, Xiang J (2005) Combined CD4+ Th1 effect and lymphotactin transgene expression enhance CD8+ Tc1 tumor localization and therapy. Gene Ther 12:999–1010
Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC (2006) A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203:1685–1691
Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y (2006) IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177:566–573
Zou W, Restifo NP (2010) T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol 10:248–256
Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea JJ (2007) Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26:371–381
Bolitho P, Voskoboinik I, Trapani JA, Smyth MJ (2007) Apoptosis induced by the lymphocyte effector molecule perforin. Curr Opin Immunol 19:339–347
Rosenberg SA, Dudley ME (2004) Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA 101(Suppl 2):14639–14645
Dudley ME, Rosenberg SA (2007) Adoptive cell transfer therapy. Semin Oncol 34:524–531
Ogawa M, Umehara K, Yu WG, Uekusa Y, Nakajima C, Tsujimura T, Kubo T, Fujiwara H, Hamaoka T (1999) A critical role for a peritumoral stromal reaction in the induction of T-cell migration responsible for interleukin-12-induced tumor regression. Cancer Res 59:1531–1538
Mukai S, Kjaergaard J, Shu S, Plautz GE (1999) Infiltration of tumors by systemically transferred tumor-reactive T lymphocytes is required for antitumor efficacy. Cancer Res 59:5245–5249
Kottke T, Sanchez-Perez L, Diaz RM, Thompson J, Chong H, Harrington K, Calderwood SK, Pulido J, Georgopoulos N, Selby P, Melcher A, Vile R (2007) Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res 67:11970–11979
Brown IE, Blank C, Kline J, Kacha AK, Gajewski TF (2006) Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol 177:4521–4529
Kline J, Brown IE, Zha YY, Blank C, Strickler J, Wouters H, Zhang L, Gajewski TF (2008) Homeostatic proliferation plus regulatory T-cell depletion promotes potent rejection of B16 melanoma. Clin Cancer Res 14:3156–3167
Nurieva R, Yang XO, Chung Y, Dong C (2009) Cutting edge: in vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J Immunol 182:2565–2568
Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C (2009) Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol 39:216–224
Zhang S, Bernard D, Khan WI, Kaplan MH, Bramson JL, Wan Y (2009) CD4+ T-cell-mediated anti-tumor immunity can be uncoupled from autoimmunity via the STAT4/STAT6 signaling axis. Eur J Immunol 39:1252–1259
Overwijk WW, Lee DS, Surman DR, Irvine KR, Touloukian CE, Chan CC, Carroll MW, Moss B, Rosenberg SA, Restifo NP (1999) Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc Natl Acad Sci USA 96:2982–2987
Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, Szeliga W, Wang Y, Liu Y, Welling TH, Elder JT, Zou W (2008) Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol 181:4733–4741
Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G (2010) Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol 184:1630–1641
He T, Zong S, Wu X, Wei Y, Xiang J (2007) CD4+ T cell acquisition of the bystander pMHC I colocalizing in the same immunological synapse comprising pMHC II and costimulatory CD40, CD54, CD80, OX40L, and 41BBL. Biochem Biophys Res Commun 362:822–828
Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA (2002) In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity 17:211–220
Purvis HA, Stoop JN, Mann J, Woods S, Kozijn AE, Hambleton S, Robinson JH, Isaacs JD, Anderson AE, Hilkens CM (2010) Low-strength T-cell activation promotes Th17 responses. Blood 116:4829–37
Xia D, Hao S, Xiang J (2006) CD8+ cytotoxic T-APC stimulate central memory CD8+ T cell responses via acquired peptide-MHC class I complexes and CD80 costimulation, and IL-2 secretion. J Immunol 177:2976–2984
Vremec D, Pooley J, Hochrein H, Wu L, Shortman K (2000) CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol 164:2978–2986
Prlic M, Hernandez-Hoyos G, Bevan MJ (2006) Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J Exp Med 203:2135–2143
Xiang J, Moyana T (1998) Cytotoxic CD4+ T cells associated with the expression of major histocompatibility complex class II antigen of mouse myeloma cells secreting interferon-gamma are cytolytic in vitro and tumoricidal in vivo. Cancer Gene Ther 5:313–320
Sas S, Chan T, Sami A, El-Gayed A, Xiang J (2008) Vaccination of fiber-modified adenovirus-transfected dendritic cells to express HER-2/neu stimulates efficient HER-2/neu-specific humoral and CTL responses and reduces breast carcinogenesis in transgenic mice. Cancer Gene Ther 15:655–666
Acknowledgments
This study was supported by a grant of Canadian Institute of Health Research (MOP 79415). Manjunatha Ankathatti Munegowda was supported by Dean’s Scholarship of University of Saskatchewan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ankathatti Munegowda, M., Deng, Y., Mulligan, S.J. et al. Th17 and Th17-stimulated CD8+ T cells play a distinct role in Th17-induced preventive and therapeutic antitumor immunity. Cancer Immunol Immunother 60, 1473–1484 (2011). https://doi.org/10.1007/s00262-011-1054-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-011-1054-y