
Abstract
Prior to the advent of VEGF-targeted therapies, renal cell carcinoma (RCC) was among the few solid tumors shown to respond to cytokine-based therapies such as interleukin-2 (IL-2) and interferon alpha. Previous work has shown that aminobisphosphonates, including zoledronic acid (ZA), are capable of activating human Vγ9 Vδ2 T cells in vitro, and these cells can be further expanded with IL-2. Moreover, these Vγ9 Vδ2 T cells have cytolytic activity in vitro to multiple human tumor cell lines. In the current report, we have conducted a pilot trial in patients with metastatic RCC, evaluating different doses of ZA in combination with low-dose IL-2 to determine whether combining these agents can promote in vivo proliferation of Vγ9 Vδ2 T cells and elicit an antitumor response. In 12 patients evaluated, no objective clinical responses were observed by RECIST criteria; however, two patients experienced prolonged stable disease. A modest increase in Vγ9 Vδ2 T-cell frequency could be detected by Day 8 of therapy in four of the nine patients who received at least one cycle of therapy, but not to the magnitude anticipated from preclinical models. Repeated administration of IL-2 and ZA resulted in both a diminished in vivo percentage of Vγ9 Vδ2 T cells as well as impaired expansion in vitro after the first cycle of therapy. These results suggest that repeated administration of IL-2 and ZA, at the doses and schedules used in this trial, may actually inhibit the proliferative capacity of Vγ9 Vδ2 T cell in patients with metastatic RCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Renal cell carcinoma (RCC) was the ninth most common cancer diagnosed in the United States during 2009, with over 58,000 new diagnoses and more than 13,000 deaths [1]. Sporadic RCC has numerous histologic subtypes that occur with varying incidences; clear cell carcinoma and variants with predominantly clear cell histology represent the majority of these tumors, and hence the most studied [2]. The mainstay of treatment for RCC is resection or ablation for localized and/or oligometastatic disease. Until 2005, the only proven treatment options for patients with unresectable or metastatic disease were immune-based therapies with interleukin-2 (IL-2) and/or interferon alpha (IFNα) [3]. Since 2005, numerous agents targeting vascular endothelial growth factor and its receptor, and agents targeting mTOR, have demonstrated efficacy in reducing RCC tumor volumes and prolonging median survival of patients over that achieved with these immune-based therapies [4–9]. This has led to five new FDA drug approvals [10]. While this has clearly changed the landscape for the treatment of RCC, and many other agents in these classes are being investigated, unfortunately despite the marked disease response rates complete responses are exceedingly rare. Thus, treatment with these agents remains chronic and costly. Consequently, there is a need for further improved treatments.
Interleukin-2 (IL-2) was the first drug approved for the treatment of metastatic RCC [11]. IL-2 is a glycoprotein that is mainly produced by helper T cells (CD4+) in response to various antigenic stimulations. While the exact mechanism of antitumor activity in humans is unknown, IL-2 release has been shown to trigger a complex cytokine network resulting in interferon alpha (IFNα) production by macrophages, differentiation of lymphokine-activated killer (LAK) cells, and enhancement of other immune cells including other T cells, B cells, and NK cells [12]. This cascade of events leads to the release of various other cytokines leading to the total immune/inflammatory reaction and resultant toxicity. A comprehensive summary of 255 renal cell cancer patients participating in 7 phase II clinical trials in which they received high-dose IL-2 described an overall response rate of 14%, in which 5% were complete and 9% were partial responses, with a median response duration of 20 months [13]. The median survival duration for all patients was 16.3 months and had not yet been reached for complete responders. Unfortunately, the toxicity of high-dose IL-2 is significant and led to the early discontinuation of treatment in approximately 5% of patients. Studies exploring different doses, schedules, and routes of administration have led to feasible treatments with similar response rates, including a small number of complete responses [14–16]. For example, the subcutaneous administration of low-dose IL-2 therapy has been shown to be effective and well tolerated in 44 patients with metastatic renal cell cancer followed over a 10-year period. An objective response rate of 22% was observed, including 2 complete (4%) and 8 (18%) partial responses, and the median duration of response was 12 months (range 4–83+ months). The 10-year survival rate was 5%, confirming clear survival prolongation in select patients [16].
Approximately 3–6% of human peripheral blood lymphocytes express the γδ T-cell receptor (TCR) [17]. The majority of these cells (e.g., 60 to >90% of γδ T cells in adults) express the Vγ9 Vδ2 TCR variable segment. Vγ9 Vδ2 T lymphocytes represent part of the innate immune system, recognizing phosphorylated nonpeptidic microbial metabolites (phosphoantigens, PAgs) and alkylamines—molecules that are structurally quite distinct from the classical antigens and superantigens interacting with αß TCRs [18, 19]. This recognition of phosphorylated molecules requires neither antigen uptake nor processing, nor MHC class I or class II expression, allowing for a very rapid response to immune challenge. Different subsets of Vγ9 Vδ2 T cells have been implicated in the primary response to infectious agents and possibly tumor cells [20]. For example, engagement of the natural killer gene 2D receptor (NKG2D) receptor on γδ T cells with the MICA antigen of epithelial tumor cell lines led to cell lysis [21, 22]. Furthermore, autologous RCC cells can be lysed by Vγ9 Vδ2 T cells in a fashion that requires both the TCR and NKG2D molecules [23]. Comparative characterization of these cells in renal tumors, normal kidney, and peripheral blood of RCC patients has shown that the Vγ9 Vδ2 T-cell subset selectively infiltrates the tumor mass [23–25] and makes up the highest percentage of all activated CD3+ T cells among the tumor infiltrating lymphocytes [26]. Kobayashi et al. [25] found that patients with renal cell carcinoma had higher numbers of circulating γδ T cells that correlated with later stages of disease. Interestingly, these numbers of circulating γδ T cells decreased following surgery and may suggest a role in both active immune response as well as immunosurveillance. Taken together, these results demonstrate that Vγ9 Vδ2 T cells have cytolytic activity against RCC, and elevated circulating levels of these cells may reflect an active tumor response in vivo.
Vγ9 Vδ2 T cells represent a small percentage of circulating lymphocytes, suggesting that expansion and activation of Vγ9 Vδ2 T cells could promote an antitumor effect on RCC. Viey et al. [23] found that when the peripheral Vγ9 Vδ2 T cells isolated from the blood of patients with RCC were stimulated and expanded with IL-2 and Phosphostim (a synthetic phosphoantigen, bromohalohydrin pyrophosphate or BrHPP), they expressed activation markers and exhibited an effector/memory phenotype. Kunzmann and colleagues reported that Vγ9 Vδ2 T cells could be stimulated in patients with multiple myeloma after treatment with the aminobisphosphonate pamidronate [27]. Previous work in nonhuman primates confirms Vγ9 Vδ2 T cells can be upregulated by in vivo administration of Phosphostim [28] or an aminobisphosphonate [29] when given in conjunction with IL-2. Zoledronic acid (ZA) is a potent aminobisphosphonate used in the treatment for hypercalcemia and bone metastases that has been shown to cause a potent expansion of Vγ9 Vδ2 T cells upon in vivo administration in multiple myeloma patients with IL-2 [30]. These results suggest that ZA can effectively activate Vγ9 Vδ2 T cells, and subsequent IL-2-induced expansion of Vγ9 Vδ2 T-cell numbers may be effective in the treatment for human malignancy.
This concept of using the combination of IL-2 with ZA to activate Vγ9 Vδ2 T cells in the treatment for both hematologic and solid tumor malignancies has been tested in several clinical trials. Dieli et al. [31] randomized eighteen patients with metastatic, castrate resistant prostate cancer to receive ZA at 4 mg with or without IL-2 given at a fixed dose SQ at 0.6 × 106 U every 21 days. Five out of nine patients in the combination therapy cohort showed sustained or increased absolute number of Vγ9 Vδ2 T cells that correlated with PSA response. This same group conducted a similar study in ten women with metastatic breast cancer treated with ZA at 4 mg IV with IL-2 given at a fixed dose SQ of 1 × 106 U every 21 days [32]. Three patients on this pilot trial were found to have an increase in circulating Vγ9 Vδ2 T cells that correlated with a decrease in serum CA15-3 levels. These patients also showed radiologic response of either stable disease or partial response. Abe et al. [33] collected peripheral blood mononuclear cells (PBMC) from six patients with refractory multiple myeloma and activated Vγ9 Vδ2 T cells ex vivo with ZA and IL-2 prior to reinfusion of these cells. All patients had some degree of immunologic response with four of the six patients showing stabilization in serum M-protein levels. These studies confirm that expansion of Vγ9 Vδ2 T cells might be a therapeutic approach for the treatment for patients with advanced malignancies.
Renal cell carcinoma is an ideal disease to examine the in vivo impact of IL-2 and ZA on Vγ9 Vδ2 T cells, in particular because IL-2 is an FDA-approved therapy for RCC and ZA is commonly used to treat hypercalcemia and bone metastases, two commonly encountered complications of RCC. To that end, we have conducted a pilot trial of IL-2 and ZA in patients with recurrent and/or metastatic RCC. The primary objective of this study was to evaluate the antitumor response rate of low-dose interleukin-2 in combination with ZA on patients with unresectable and/or metastatic RCC. Therefore, the primary endpoint of the study was objective tumor response as defined by RECIST guidelines. The primary laboratory objective was to evaluate the effect of treatment on the frequency and activity of circulating Vγ9 Vδ2 T cells. Secondary clinical objectives included progression-free survival, overall survival, and toxicity evaluation.
Materials and methods
Patient population and study design
A single institution, open label, pilot trial was conducted at the University of Wisconsin Carbone Cancer Center. Patients with recurrent, metastatic renal cell cancer with clear cell, or predominant clear cell, histology were offered enrollment between 2003 and 2006. Patients with sarcomatoid variant cancers or collecting duct malignancies were excluded given historically poor response rates with IL-2-based therapies. Patients were required to have measurable disease, defined as at least one lesion that could be accurately measured in at least one dimension. Patients who had received prior cytokine, chemotherapy, hormonal, or other immune-based (including vaccine- or cellular-based) therapies for their renal cancer were not eligible. In addition, patients with a prior use of IV bisphosphonates were not eligible. Patients were required to have an ECOG performance status of 0 or 1 at the time of enrollment and normal liver, cardiac, and marrow function. All patients signed informed consent prior to enrollment approved by the University of Wisconsin Institutional Review Board. Dose modifications and delays were based on observed toxicities and laboratory parameters. The CTC (NCI Common Terminology Criteria) version 2.0 was used for toxicity and adverse event reporting. In the absence of treatment delays due to adverse events, treatment was continued until disease progression, intercurrent illness preventing further administration of treatment, unacceptable adverse events, patient withdrawal, or investigator determination that other therapies were warranted.
Treatment plan
The initial study design treated patients with ZA at a starting dose of 4 mg intravenously on Day 1 of each 28-day cycle and IL-2 at a starting dose of 7 × 106 U/m2/day by subcutaneous injection Days 1–5, weekly in weeks 1 through 3 of each cycle, with standard premedication with oral acetaminophen (Fig. 1). Based on observed toxicities in Patients 1–6, and concern that the dose of IL-2 may be too high to permit expansion of Vγ9 Vδ2 T cells (as preliminary studies in the initial patients failed to result in a significant augmentation of Vγ9 Vδ2 T cells), IL-2 was reduced to a dose of 1 × 106 U/m2/day by subcutaneous injection Days 1–5, on weeks 1 through 3, in 4 week (28 days) cycles. After the 9th patient, the protocol was further amended to allow for intrapatient dose escalation of ZA given concerns that the dose of ZA might be too low or too high to sufficiently augment Vγ9 Vδ2 T cells. While receiving protocol therapy, patients were recommended to take an oral calcium supplement of at least 500 mg and a multi-vitamin containing 400 IU of vitamin D daily.

Treatment Schema. Patients enrolled on this study were treated on Day 1 (D1) of week 1 with IL-2 and ZA, with dosing shown in Table 2. IL-2 was administered subcutaneously daily from Days 1–5, 8–12, and 15–19 of a 28-day course
Assessments, follow-up, and radiographic monitoring
Patients were assessed on Day 1 of each cycle with a medical history, physical examination, and blood work including CBC with differential chemistries and liver function tests. Weekly safety laboratories, including CBC, LFTs, and creatinine, were checked prior to administration of IL-2. Any abnormal values (from patient’s baseline) required repeat testing until re-treatment criteria were met or the patient was removed from the study. Supportive measures were administered as needed. Disease assessments were performed with CT scans of the chest/abdomen/pelvis after every 2 cycles of therapy. Response and disease progression were based on RECIST 1.0 guidelines.
Immune response evaluation
Flow cytometric analysis
PBMC were isolated by density gradient centrifugation over Ficoll-Paque using standard techniques and stained with the following fluorochrome-labeled monoclonal antibodies for respective experiments: Vγ9-FITC, Vδ2-PE, CD3-FITC, αβ TCR-PE, CD4-APC, CD8-PE, Vδ2-FITC, CD27-PE, CD45RA-APC (Beckton Dickinson (BD) Bioscience Pharmingen, San Jose, CA), or isotype controls (IgG1-FITC, IgG1-PE (Sigma, St. Louis, MO), IgG1-APC (BD)). After labeling, the cells were washed and fixed with paraformaldehyde (Sigma, St. Louis, MO) and the samples analyzed on a FACSCalibur (Becton–Dickinson Biosciences, Mountain View, CA). At least 20,000 events were collected for all samples and analyzed using FlowJo software (TreeStar, San Carlos, CA).
Vγ9 Vδ2 T-cell Proliferation Analysis
PBMC isolated from patients were cultured at 37°C/5%CO2 in 96-well sterile microtiter plates at 5 × 105 PBMC/well in complete RPMI-1640 culture medium with IL-2 at 200 U/ml (Boehringer Mannheim, Germany) and twofold serial dilutions of zoledronic acid (ZA, Zometa®, Novartis Pharmaceuticals), starting from 16 μM ZA to 0.06 μM ZA. On Day 7 of culture, the frequency of Vγ9 Vδ2 T cells was determined by flow cytometry, as above. The expansion index was calculated as the absolute number of Vγ9 Vδ2 cells per culture on Day 7 divided by the absolute number of Vγ9 Vδ2 cells seeded in culture on Day 0.
Results
Patient characteristics
Between 2003 and 2006, this pilot trial enrolled twelve patients with recurrent, metastatic RCC with clear cell, or predominantly clear cell, histology. All patients initially had an ECOG performance status of 0 or 1. As shown in Table 1, the majority of these patients had Memorial Sloan Kettering Cancer Center (MSKCC) risk status of intermediate. The three patients with a risk categorization of poor did not complete cycle one of therapy due to toxicity and/or rapid disease progression. The majority had also undergone nephrectomy, and none had received prior immunotherapy such as IL-2 or interferon, or treatment with aminobisphosphonates. All patients had metastatic disease, with nearly all patients having pulmonary lesions. Other common sites of metastases included liver, lymph node, bone, and adrenal gland. Only three had received prior systemic therapy consisting of investigational agents, of which two were VEGF-targeted therapies.
Study conduct and adverse events
Overall, the majority of patients experienced predominantly Grade 1/2 toxicities as detailed in Table 2. The most common Grade 1/2 toxicities observed included fever, chills, and injection-site reactions. Patient 4 experienced a Grade 4 myocardial infarction that was believed unrelated to study medications. Patient 5 was enrolled but did not receive any study medications due to rapid progression of disease. Patient 6 discontinued treatment prior to completion of Cycle 1 secondary to a Grade 4 elevation in creatinine that resolved to baseline levels within 3 weeks off of treatment. Given the Grade 4 events and the desire to limit potential renal toxicity given its association with both IL-2 and ZA, the dose of IL-2 was decreased to 1 × 106 U/m2/day on the same dosing schedule for all subsequent patients. Patient 8 came off study prior to completion of Cycle 1 due to intolerable, Grade 2 fatigue, and Grade 2 myalgias/arthralgias. Other common reactions included influenza-like symptoms after infusion of ZA and injection-site reactions from IL-2.
Fewer adverse events were observed at the lower dose of IL-2, and hence, there were no safety concerns for this dose in combination with zoledronic acid; however, on the basis of results from in vitro immunologic analyses discussed below, the trial was amended for Patients 10–12 to begin at a lower dose of ZA and escalate the dose after each cycle of therapy, to specifically promote in vivo expansion of Vγ9 Vδ2 T cells. The starting dose of ZA in these individuals was 400mcg IV on Day 1 of Cycle 1 and escalated to a maximum dose of 3 mg by Cycle 5 and for all subsequent cycles. Dosing for individual patients is described in detail in Table 3, with Patients 1–9 subsequently described as Cohort A and Patients 10–12 as Cohort B. A single Grade 3 toxicity of hyperglycemia was observed in Patient 10 in Cohort B, with no other Grade 3 or 4 toxicities observed in Cohort B. Grade 1 and 2 toxicities were similar to those observed in Cohort A (Table 2).
Clinical response
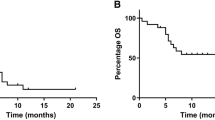
As seen in Table 3, no partial or complete responses were observed. Five of the eight patients who received more than one cycle of therapy had radiographically stable disease, not meeting criteria for partial response or progression, with a median duration of 7 months (range 4–33 months). These patients included a range of MSKCC risk status categorization from favorable to intermediate. None of the three patients with a poor MSKCC risk status completed even one cycle of therapy due to disease progression and/or toxicity.
Immune response evaluation
In Cohort A, lymphocytes isolated from these patients were analyzed by flow cytometry for the frequency of T-cell subsets, including Vγ9 Vδ2 T cells, with a representative experiment shown in Fig. 2. As shown in Fig. 3, the percentage of both CD3+ T cells and Vγ9 Vδ2 T cells declined by Day 4. Subsequently lowering the dose of IL-2 in Patients 8 and 9 abrogated the loss of CD3+ T lymphocytes but did not impact Vγ9 Vδ2 T cells. Expansion of Vγ9 Vδ2 T cells was observed in patients on Day 1 of Cycle 2 in Patients 1–3, suggesting that the kinetics of expansion were occurring sometime after Day 4. We then began collecting PBMC samples on Day 8 for Patients 8 and 9, showing significant expansion of Vγ9 Vδ2 T cells but not overall CD3+ T lymphocytes. Interestingly, Patient 9 showed an expansion of Vγ9 Vδ2 T cells by Day 8 of Cycle 1 that persisted to Day 1 of Cycle 2. However, the percentage of Vγ9 Vδ2 T cells as well as the capacity for in vivo expansion fell by Cycle 3 of therapy and did not recover to baseline levels, suggesting a possible loss of these cells with subsequent treatment.

Representative example demonstrating the effect of ZA and IL-2 therapy on Vγ9 Vδ2 T cells in RCC patients. PBMC were collected from patients with RCC before and after ZA and IL-2 therapy and stained with Vγ9-FITC- and Vδ2-PE-labeled antibodies. Lymphocytes were isolated by forward and side scatter then gated by FITC and PE to identify Vγ9 Vδ2 T cells. The dual parameter scatter plots shown here are representative examples using lymphocytes from Patient 7 to demonstrate the changes in Vγ9 Vδ2 T cells on Days 0, 4, and 8 of Cycles 1 and 2. The percentage of Vγ9 Vδ2 cells among gated lymphocytes is shown

CD4+ and CD8+ T lymphocytes do not show in vivo proliferation during ZA and IL-2 therapy in RCC patients, while Vγ9 Vδ2 T lymphocytes show minimal expansion in Cohort A. a PBMC collected from patients in Cohort A before and after ZA and IL-2 therapy were surface stained with CD4-FITC- and CD8-PE-specific surface antibodies. Lymphocytes were isolated by forward and side scatter and then gated by CD3-FITC to determine the T-cell subset. The CD3-FITC-positive population was further analyzed for the expression of CD4-APC and CD8-PE to define these T-cell subsets. Results illustrate percentage of peripheral CD4+ and CD8+ lymphocytes in Patients 1–4 and 7. b PBMC collected from patients in Cohort A before and after ZA and IL-2 therapy and stained with Vγ9-FITC- and Vδ2-PE-labeled antibodies. Lymphocytes were isolated by forward and side scatter and then gated by FITC and PE to isolate Vγ9 Vδ2 T cells. Blood samples were drawn pretreatment on Day 1 (designated as Day 0 as pretreatment) and post-treatment on Day 4 of each cycle, i.e., C1D0, C1D4, C2D0, and C2D4. C Cycle, D Day
While in vivo expansion was observed in a few patients, the magnitude of response was not to the degree expected from prior studies in nonhuman primates [29]. In addition, fewer Vγ9 Vδ2 T cells were observed in patients with each subsequent cycle, suggesting a loss of responsiveness to treatment, or potentially a loss or sequestration of Vγ9 Vδ2 T cells, occurring over time. To explore potential mechanisms of these findings, we evaluated the proliferative ability of these Vγ9 Vδ2 T cells isolated from the peripheral blood of these patients at the different time points. The aim was to determine the optimal dose of ZA for in vitro stimulation of Vγ9 Vδ2 T cells and thus to potentially guide in vivo dosing and also to evaluate the functional activity of these cells following treatment. PBMC from Patients 1–4 and 7 in Cohort A were cultured in vitro with IL-2 at 200 U/ml and increasing concentrations of ZA as shown in Fig. 4. Culture medium with only IL-2 was used as a control. After 7 days of incubation, the frequency of Vγ9 Vδ2 T cells was assessed by flow cytometry. We found that the expansion of Vγ9 Vδ2 T cells for all patients, except Patient 1, showed maximal in vitro expansion at a concentration of 0.5–1 μM ZA, whereas higher doses led to less proliferation of these cells (Fig. 4). This is similar to previously published studies [34, 35] where 1 μM ZA demonstrated optimal in vitro Vγ9 Vδ2 T-cell proliferation. In all the patients, IL-2 alone induced minimal expansion of Vγ9 Vδ2 T cells (data not shown). In addition, we observed that, in general, Vγ9 Vδ2 T cells demonstrated maximal expansion on C1D1 for most of the patients, followed by a diminished proliferative response after exposure to ZA and IL-2 on the other days of the cycle and in subsequent cycles. This observation was most apparent in Patients 2 and 7 (Fig. 4). Because proliferative capacity of Vγ9 Vδ2 T cells is a property of the naïve and central memory cell subset, we then examined these subsets in Vδ2 T cells based on CD27 and CD45RA surface marker expression as shown in Fig. 5. As shown for a single patient (#9), we observed a transient decrease in the percentage of naïve Vδ2 T cells and increase in the effector memory subset by Day 8 after treatment. Overall, the frequency of naïve and central memory cells did not return to pretreatment levels by Day 16 (Fig. 5), which were diminished in samples obtained at later time points in other patient samples (data not shown). Taken together, these results suggested that ZA and IL-2 might be affecting the maturation of these cells and/or not permitting the adequate recovery of Vδ2 T cells with proliferative capacity.

In vitro expansion of Vγ9 Vδ2 T cells from RCC patients in response to ZA and IL-2. PBMC from Patients 1–4 and 7 in Cohort A were cultured with the indicated concentration of ZA in the presence of 200 U/ml IL-2. Absolute numbers of viable Vγ9 Vδ2 T cells were calculated by flow cytometry after 7 days in culture. Graphs show the expansion of Vγ9 Vδ2 T cells from Patients 1–4 and 7, respectively. Expansion indices indicate the ratio between Vγ9 Vδ2 T cells on Day 7 and the initial number of Vγ9 Vδ2 T cells seeded in culture

Vδ2 T cells show increased differentiation toward an effector memory subset after treatment with IL-2 and ZA with a relative decrease in the naïve subset. PBMC were collected from Patient 9 before and after cycle one of ZA and IL-2 therapy at Days 0, 8, and 16. Lymphocytes were isolated by forward and side scatter and then gated by Vδ2 expression (left panels). Vδ2 T-cell subsets were then evaluated for CD27 and CD45RA surface marker expression (right panels), and the percentages of Vδ2 T-cell subsets for naïve, effector, central memory and effector memory cells (with gates set by IgG controls) are shown in the quadrants
The in vitro studies further suggested that concentrations of ZA in the low micromolar range could be optimal for inducing expansion of Vγ9 Vδ2 T cells. Therefore, doses of ZA that were too high or too low may not have permitted optimal expansion and might have negatively impacted the ability of these cells to proliferate upon subsequent exposure to ZA. Consequently, for Patients 10–12 (Cohort B), the study was modified to permit an intrapatient dose escalation using a starting ZA dose of 400 μg on Day 1 of Cycle 1, with an increase in the ZA dose for subsequent cycles. As shown in Fig. 6, the percentage of CD3+ T lymphocytes was relatively consistent from cycle to cycle in this cohort. A small percentage increase in Vγ9 Vδ2 T cells was detected in Patients 10 and 12 by Days 8 and 4, respectively. However, this was again not nearly the degree of expansion expected based on data from nonhuman primates [28]. Similar to Patient 9 from Cohort A, a subsequent loss of Vγ9 Vδ2 T cells was observed with each subsequent cycle of therapy. Based on the inability to promote a sustained in vivo expansion of Vγ9 Vδ2 T cells at the doses and schedules of IL-2 and ZA shown, and the absence of objective disease responses observed, this study was closed to further accrual.

CD4+ and CD8+ T lymphocytes do not show in vivo proliferation during ZA and IL-2 therapy in RCC patients, while Vγ9 Vδ2 T lymphocytes show minimal expansion in Cohort B. a PBMC collected from patients in Cohort B before and after ZA and IL-2 therapy were evaluated for CD4 and CD8 expression as in Fig. 3. b PBMC collected from patients in Cohort B before and after ZA and IL-2 therapy were evaluated for Vγ9 Vδ2 T-cell frequencies as in Fig. 3. C Cycle, D Day
Discussion
We report the results of a pilot trial of zoledronic acid and IL-2 in patients with recurrent/metastatic renal cell carcinoma of clear cell or predominant clear cell histology to promote the in vivo expansion of Vγ9 Vδ2 T cells. The primary clinical objective of this trial was objective radiologic response based on RECIST 1.0 guidelines. The primary laboratory objective of this trial was to evaluate the effect of treatment on circulating Vγ9 Vδ2 T cells with the goal of promoting an in vivo expansion of these cells and a resulting antitumor immune response. We have found that the combination of ZA with low-dose IL-2 administered subcutaneously was relatively well tolerated with similar toxicities to those previously reported with IL-2 alone. The best response to therapy was stable disease in five of the nine patients who received at least one cycle of therapy. However, this combination of agents at the doses evaluated was not successful in augmenting Vγ9 Vδ2 T cells in vivo, rather the proliferative capacity of these cells was found to diminish with treatment. These findings suggest that (1) the combination of IL-2 and ZA in the doses evaluated has a similar toxicity profile to single-agent IL-2 in patients with advanced RCC; (2) this combination of agents did not sufficiently expand Vγ9 Vδ2 T cells in vivo to promote a potential antitumor response in patients with advanced RCC; and (3) repeated in vivo stimulation of Vγ9 Vδ2 T cells with ZA and IL-2 at these doses ultimately lead to a loss of proliferative capacity of these cells. These results suggest that in vivo expansion of Vγ9 Vδ2 T cells with IL-2 and ZA may be difficult to achieve in patients with metastatic RCC at the typical doses and schedules used for these agents and that repeated stimulation may actually blunt the proliferative capacity of these cells, potentially by affecting the maturation of naïve Vγ9 Vδ2 T cells.
Prior to the advent of targeted therapies, the mainstay of treatment for advanced RCC was cytokine therapy with IL-2 or IFNα [3]. Due to commonly encountered sequelae of hypercalcemia and bone metastases, bisphosphonates are frequently used in this patient population. When combined in the treatment for patients with intermediate or high risk RCC in this trial, the best clinical response was stable disease in five patients with prolonged disease control in two of these patients up to 280 and 224 days. These results suggest that the addition of ZA did not significantly enhance the efficacy of IL-2 in patients with advanced RCC, although this was a very small trial. Significant toxicities were observed in this trial, with Grade 4 toxicities of myocardial infarction, elevated creatinine, and hyperglycemia. Grade 3 toxicities of noncardiac chest pain were observed 25% of patients and hypocalcemia in 25% of patients. Other Grade 1/2 toxicities included renal toxicity, fatigue, nausea, and pain. In addition, two of the twelve patients enrolled on this study did not complete the first cycle due to treatment related toxicities. The dose of IL-2 in this pilot trial was reduced for Patients 7–12 due to concerns of overlapping toxicities in regards to renal function, and fewer toxicities >Grade 2 were observed, suggesting that the majority of the adverse events observed were due to IL-2. The nature of adverse events was similar to other trials using subcutaneous administration of IL-2 [36].
The primary laboratory objective in this trial was to promote in vivo expansion of Vγ9 Vδ2 T cells via stimulation by ZA and growth factor support by IL-2. In cohort A, none of the first six patients enrolled on study exhibited an expansion of Vγ9 Vδ2 T cells at Day 4. We then began collecting samples at Day 8, which revealed expansion in Patients 8 and 9, while in Patient 7, the numbers of Vγ9 Vδ2 T cells recovered by Day 8. This decline in cell numbers after initial exposure to ZA and IL-2 has been seen in previous studies and attributed to possible activation-induced transmigration Vγ9 Vδ2 T cells through the endothelial layer [37]. These Vγ9 Vδ2 T cells have been observed to reappear several days later in an activated state with increase in serum IFN-γ detection [37]. Other potential etiologies of these findings include a decline in cell numbers due to activation-induced cell death or general toxicity of the drug regimen. These results contrast to those found in nonhuman primates, where treatment with a synthetic phosphoantigen and IL-2 resulted in a dramatic increase in circulating Vγ9 Vδ2 T cells, up to 80% of circulating CD3+ T cells [28]. Prolongation of IL-2 administration beyond 7 days did not further enhance this response. It is also possible that the dose of ZA was not appropriate to permit expansion of Vγ9 Vδ2 T cells in the periphery. This is supported by our in vitro proliferation studies, which demonstrated maximal expansion at intermediate doses of 0.5–1 μM, with decreased responsiveness at both lower and higher ZA doses. While the pharmacokinetics of ZA were not collected in this study, an analysis of plasma concentrations of ZA after a single infusion of 4 mg of ZA found that peak plasma concentration at the end of a 15-min infusion was 264 ± 86 ng/mL (~0.91 μM) [38], within the range of ZA dose expected to promote expansion of Vγ9 Vδ2 T cells. Given safety concerns about increasing the dose of ZA above 4 mg, and to address the possibility that the dose of ZA might have been too high, the protocol was amended for Patients 10–12 to receive ZA in a dose escalation format. Despite this dose escalation design, we did not observe a greater in vivo proliferation in circulating Vγ9 Vδ2 T cells after cycle one of therapy, suggesting that Vγ9 Vδ2 T-cell expansion was likely not impacted by the dose of ZA. A previous report by Dieli et al. treated patients with metastatic prostate cancer with a single, fixed dose of IL-2 at 0.6 × 106 U and ZA at 4 mg IV on Day 1 of each 21-day cycle [31]. This study found increased levels of circulating Vγ9 Vδ2 T cells at 3, 6, and 9 months that appeared to correlate with disease response. Similar results were reported in patients with metastatic breast cancer [32]. As subcutaneous administration of IL-2 has been shown to improve disease control, the current study was designed to provide an IL-2 dose that would still retain primary antitumor activity. However, our results suggest that the dose of IL-2 and frequency of administration in this trial was disadvantageous based on the lack of prolonged γδ T-cell response.
The laboratory endpoints also included assessment of in vitro proliferation of Vγ9 Vδ2 T cells after multiple cycles of treatment with IL-2 and ZA. In Patients 1–4 and 7, in vitro proliferation assays demonstrated maximal proliferation of Vγ9 Vδ2 T cells at optimal ZA doses prior to exposure to ZA and IL-2 as discussed above. However, Vγ9 Vδ2 T cells showed decreasing proliferation in response to stimulation in vitro that continued to fall with each subsequent cycle. These results suggest that the proliferative capacity of Vγ9 Vδ2 T cells can be exhausted during in vivo expansion. Sicard et al. [28] found similar results in nonhuman primates, wherein the amplitude of the Vγ9 Vδ2 T-cell response in nonhuman primates decreased with each subsequent cycle of IL-2 and the synthetic phosphoantigen. This phenomenon has also been observed by Dieli et al. in patients with prostate cancer. However, these authors found that despite lowered proliferative capacity, these cells secreted a higher concentration of IFNγ at the same time points [31]. This may be explained by TCR downregulation and by the type of γδ T cells involved in proliferation and IFNγ production, as proliferative activity of Vγ9 Vδ2 T cells is a property of the naïve and central memory cell subset, whereas expression of IFNγ is a property of effector memory cells [39]. As seen in Fig. 5, a dramatic increase in the effector memory subset was observed by Day 8 in Patient 9, that declined by Day 16. Further studies in RCC patients could further explore the specific subsets of Vγ9 Vδ2 T cells during therapies along with measuring IFNγ production to determine the response of the effector subset to ZA and IL-2 therapy. This may improve our understanding how different Vγ9 Vδ2 T-cell subsets could contribute to clinical responses.
While the results of this trial do not support the use of IL-2 and ZA at the doses evaluated for in vivo expansion of Vγ9 Vδ2 T cells in the treatment for advanced RCC, other groups have utilized adoptive transfer of these cells to directly evaluate their antitumor activity. Kobayashi et al. [40] have demonstrated both safety and preliminary evidence of antitumor effect when autologous γδ T cells were activated in vitro with IL-2 and a phosphoantigen and then infused into patients with advanced RCC. A later case report from this group used this product in combination with low-dose IL-2 and ZA to treat a patient with metastatic RCC [41]. A traditional Phase I trial with an autologous cell therapy was used in combination with a seven-day course of low-dose IL-2 in patients with metastatic RCC [42]. This product was produced by collecting PBMC from patients and stimulating with BrHPP at 3 μM and IL-2 at 20 ng/mL on Day 1 and 60 ng/mL on Days 4–14 of culture. After 15 days of culture, this product enriched for Vγ9 Vδ2 T cells was infused into the donor. IL-2 at 2 × 106 U/m2/day was given subcutaneously for 7 days, beginning with Day 1 of Cycle 2. Interestingly, peripheral Vγ9 Vδ2 T cells were initially cleared from circulation but reappeared at the end of IL-2 administration again raising the question of whether these cells undergo AICD, tumor migration, or other phenomena as discussed above. Evidence of disease stabilization in six of the ten patients treated on this trial suggests these cells may have a direct antitumor effect. Clinical trials to further test this adoptive transfer approach are currently underway.
In summary, this pilot trial assessed the ability of IL-2 and ZA to induce Vγ9 Vδ2 T-cell proliferation in vivo as a treatment for metastatic RCC. Adverse events were typical of those seen with IL-2 monotherapy. No complete or partial responses were observed, although 5/12 patients experienced prolonged stable disease. The doses used in this study failed to induce proliferation of Vγ9 Vδ2 T cells in vivo and led to a marked decrease in the proliferative capacity of these cells in vitro with subsequent cycles of therapy. The inability of the doses and schedules evaluated here to effectively augment these populations in vivo may be the reason we saw no significant antitumor activity. Further investigations, such as decreasing the dose or frequency of IL-2 administration, would further contribute to the approaches that seek to therapeutically augment Vγ9 Vδ2 T cells. The results of this study may guide the design of future clinical trials using bisphosphonate-stimulated Vγ9 Vδ2 T cells in cancer patients.
References
Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60(5):277–300
Linehan WM, Zbar B, Bates BE, Zelefsky MJ, Yang JC (2001) Cancer of the kidney and ureter. In: DeVita VTHS, Rosenberg SA (eds) Cancer principles and practice of oncology, 6th edn. Lippincott Williams & Wilkins, Philadelphia, pp 1362–1396
Cohen HT, McGovern FJ (2005) Renal-cell carcinoma. N Engl J Med 353(23):2477–2490
Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, Zarba JJ, Chen M, McCann L, Pandite L, Roychowdhury DF, Hawkins RE (2010) Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 28(6):1061–1068
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, Garcia-del-Muro X, Sosman JA, Solska E, Wilding G, Thompson JA, Kim ST, Chen I, Huang X, Figlin RA (2009) Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 27(22):3584–3590
Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T, Lustgarten S, Moore L, Motzer RJ (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356(22):2271–2281
Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S, Ravaud A, Golding S, Jethwa S, Sneller V (2010) Phase III Trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 28(13):2144–2150
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM (2007) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356(2):125–134
Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372(9637):449–456
Lang JM, Harrison MR (2010) Pazopanib for the treatment of patients with advanced renal cell carcinoma. Clin Med Insights Oncol 4:95–105
Motzer RJ, Mazumdar M, Bacik J, Russo P, Berg WJ, Metz EM (2000) Effect of cytokine therapy on survival for patients with advanced renal cell carcinoma. J Clin Oncol 18(9):1928–1935
Malaguarnera M, Ferlito L, Gulizia G, Di Fazio I, Pistone G (2001) Use of interleukin-2 in advanced renal carcinoma: meta-analysis and review of the literature. Eur J Clin Pharmacol 57(4):267–273
RS Fisher RI, Fyfe G (2000) Long-term survival update for high-dose recombinant interleukin-2 in patients with renal cell carcinoma. Cancer J Sci Am 6(Suppl 1):S55–S57
Amato RJ, Morgan M, Rawat A (2006) Phase I/II study of thalidomide in combination with interleukin-2 in patients with metastatic renal cell carcinoma. Cancer 106(7):1498–1506
Amato RJ, Malya R, Rawat A (2008) Phase II study of combination thalidomide/interleukin-2 therapy plus granulocyte macrophage-colony stimulating factor in patients with metastatic renal cell carcinoma. Am J Clin Oncol 31(3):237–243
Lissoni P, Bordin V, Vaghi M, Fumagalli L, Bordoni A, Mengo S, Bucovec R, Fumagalli E, Malugani F, Ardizzoia A, Giani L, Gardani GS, Tancini G (2002) Ten-year survival results in metastatic renal cell cancer patients treated with monoimmunotherapy with subcutaneous low-dose interleukin-2. Anticancer Res 22(2B):1061–1064
Lanier LL, Ruitenberg J, Bolhuis RL, Borst J, Phillips JH, Testi R (1988) Structural and serological heterogeneity of gamma/delta T cell antigen receptor expression in thymus and peripheral blood. Eur J Immunol 18(12):1985–1992
Wallace M, Malkovsky M, Carding SR (1995) Gamma/delta T lymphocytes in viral infections. J Leukoc Biol 58(3):277–283
Malkovsky M, Fisch P, Mackenzie D, Bartz SR, Radtke BE, Wallace M, Manning J, Colizzi V, Pauza CD (1992) Specificity and function of gamma delta T lymphocytes. Folia Biol (Praha) 38(5):293–306
Kato Y, Tanaka Y, Miyagawa F, Yamashita S, Minato N (2001) Targeting of tumor cells for human gammadelta T cells by nonpeptide antigens. J Immunol 167(9):5092–5098
Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T (1999) Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A 96(12):6879–6884
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285(5428):727–729
Viey E, Fromont G, Escudier B, Morel Y, Da Rocha S, Chouaib S, Caignard A (2005) Phosphostim-activated gamma delta T cells kill autologous metastatic renal cell carcinoma. J Immunol 174(3):1338–1347
Olive C, Nicol D, Falk MC (1997) Characterisation of gamma delta T cells in renal cell carcinoma patients by polymerase chain reaction analysis of T cell receptor transcripts. Cancer Immunol Immunother 44(1):27–34
Kobayashi H, Tanaka Y, Yagi J, Toma H, Uchiyama T (2001) Gamma/delta T cells provide innate immunity against renal cell carcinoma. Cancer Immunol Immunother 50(3):115–124
Kowalczyk D, Skorupski W, Kwias Z, Nowak J (1996) Activated gamma/delta T lymphocytes infiltrating renal cell carcinoma. Immunol Lett 53(1):15–18
Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M (2000) Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood 96(2):384–392
Sicard H, Ingoure S, Luciani B, Serraz C, Fournie JJ, Bonneville M, Tiollier J, Romagne F (2005) In vivo immunomanipulation of V gamma 9 V delta 2 T cells with a synthetic phosphoantigen in a preclinical nonhuman primate model. J Immunol 175(8):5471–5480
Casetti R, Perretta G, Taglioni A, Mattei M, Colizzi V, Dieli F, D’Offizi G, Malkovsky M, Poccia F (2005) Drug-induced expansion and differentiation of V gamma 9 V delta 2 T cells in vivo: the role of exogenous IL-2. J Immunol 175(3):1593–1598
Dieli F, Gebbia N, Poccia F, Caccamo N, Montesano C, Fulfaro F, Arcara C, Valerio MR, Meraviglia S, Di Sano C, Sireci G, Salerno A (2003) Induction of gammadelta T-lymphocyte effector functions by bisphosphonate zoledronic acid in cancer patients in vivo. Blood 102(6):2310–2311
Dieli F, Vermijlen D, Fulfaro F, Caccamo N, Meraviglia S, Cicero G, Roberts A, Buccheri S, D’Asaro M, Gebbia N, Salerno A, Eberl M, Hayday AC (2007) Targeting human gamma delta T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res 67(15):7450–7457
Meraviglia S, Eberl M, Vermijlen D, Todaro M, Buccheri S, Cicero G, La Mendola C, Guggino G, D’Asaro M, Orlando V, Scarpa F, Roberts A, Caccamo N, Stassi G, Dieli F, Hayday AC (2010) In vivo manipulation of Vgamma9Vdelta2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin Exp Immunol 161(2):290–297
Abe Y, Muto M, Nieda M, Nakagawa Y, Nicol A, Kaneko T, Goto S, Yokokawa K, Suzuki K (2009) Clinical and immunological evaluation of zoledronate-activated Vgamma9gammadelta T-cell-based immunotherapy for patients with multiple myeloma. Exp Hematol 37(8):956–968
Sato K, Kimura S, Segawa H, Yokota A, Matsumoto S, Kuroda J, Nogawa M, Yuasa T, Kiyono Y, Wada H, Maekawa T (2005) Cytotoxic effects of gammadelta T cells expanded ex vivo by a third generation bisphosphonate for cancer immunotherapy. Int J Cancer 116(1):94–99
Mariani S, Muraro M, Pantaleoni F, Fiore F, Nuschak B, Peola S, Foglietta M, Palumbo A, Coscia M, Castella B, Bruno B, Bertieri R, Boano L, Boccadoro M, Massaia M (2005) Effector gammadelta T cells and tumor cells as immune targets of zoledronic acid in multiple myeloma. Leukemia 19(4):664–670
Tourani JM, Lucas V, Mayeur D, Dufour B, DiPalma M, Boaziz C, Grise P, Varette C, Pavlovitch JM, Pujade-Lauraine E, Larregain D, Ecstein E, Untereiner M, Vuillemin E, Merran S, Andrieu JM (1996) Subcutaneous recombinant interleukin-2 (rIL-2) in out-patients with metastatic renal cell carcinoma. Results of a multicenter SCAPP1 trial. Ann Oncol 7(5):525–528
Wilhelm M, Kunzmann V, Eckstein S, Reimer P, Weissinger F, Ruediger T, Tony HP (2003) Gammadelta T cells for immune therapy of patients with lymphoid malignancies. Blood 102(1):200–206
Zoledronic Acid [Package Insert]. Novartis pharmaceuticals. East Hanover, NJ (2001)
Caccamo N, Meraviglia S, Ferlazzo V, Angelini D, Borsellino G, Poccia F, Battistini L, Dieli F, Salerno A (2005) Differential requirements for antigen or homeostatic cytokines for proliferation and differentiation of human Vgamma9Vdelta2 naive, memory and effector T cell subsets. Eur J Immunol 35(6):1764–1772
Kobayashi H, Tanaka Y, Yagi J, Osaka Y, Nakazawa H, Uchiyama T, Minato N, Toma H (2007) Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: a pilot study. Cancer Immunol Immunother 56(4):469–476
Kobayashi H, Tanaka Y, Shimmura H, Minato N, Tanabe K (2010) Complete remission of lung metastasis following adoptive immunotherapy using activated autologous gammadelta T-cells in a patient with renal cell carcinoma. Anticancer Res 30(2):575–579
Bennouna J, Levy V, Sicard H, Senellart H, Audrain M, Hiret S, Rolland F, Bruzzoni-Giovanelli H, Rimbert M, Galea C, Tiollier J, Calvo F (2010) Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vgamma9Vdelta2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol Immunother 59(10):1521–1530
Acknowledgments
We would like to thank Beth A. Fredricks for her invaluable technical assistance. This work was supported by an investigator-initiated award from Novartis Pharmaceuticals and by the University of Wisconsin Carbone Cancer Center Core Grant P30 CA014520. Dr. Lang is supported by NIH grant T32 CA009614.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lang, J.M., Kaikobad, M.R., Wallace, M. et al. Pilot trial of interleukin-2 and zoledronic acid to augment γδ T cells as treatment for patients with refractory renal cell carcinoma. Cancer Immunol Immunother 60, 1447–1460 (2011). https://doi.org/10.1007/s00262-011-1049-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-011-1049-8