
Abstract
Despite recent progress in the therapeutic approach of malignant haemopathies, their prognoses remain frequently poor. Immunotherapy offers an alternative of great interest in this context but defect or abnormal expression of human leukocyte antigens (HLA), frequently observed in cancer cells, limits its efficiency. Natural killer (NK) cells, which are able to kill target cells in a HLA-independent way, represent a novel tool in the treatment of haematological malignancies. Abnormal NK cytolytic function is observed in all the haematological malignancies studied, such as acute leukaemia, myelodysplastic syndromes or chronic myeloid/lymphoid leukaemia. Several mechanisms are involved in the alterations of NK cytotoxicity: decreased expression of activating receptors, increased expression of inhibitory receptors or defective expression of NK ligands on target cells. Further studies are needed to identify how each type of haematological malignancy escapes from the innate immune response. Attempts to increase the expression of activating receptors, to counteract inhibitory receptors expression, or to increase NK cell cytotoxic capacities could overcome tumour escape from innate immunity. These therapies are based on monoclonal antibodies or culture of NK cells in presence of cytokines or dendritic cells. Moreover, many novel drugs used in haematological malignancies [tyrosine kinase inhibitors, IMIDs®, proteasome inhibitors, demethylating agents, histone deacetylase inhibitors (HDACis), histamine dihydrochloride] display interesting immunomodulatory properties that affect NK cells. These data suggest that combined modalities associating cytotoxic drugs with innate immunity modulators may represent a major breakthrough in tumour eradication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite recent progress in approach of malignant haemopathies, their prognosis frequently remains poor due to the difficulty in achieving complete remission (CR) and to the high risk of relapse. Immunotherapy could thus be of great interest in this setting. Specific immunotherapy is mainly challenged by the defect of expression of human leukocyte antigens (HLA) molecules frequently observed in cancer cells, together with the progressive selection of cancer clones that have lost their HLA molecules and thus escape from immune control by specific T lymphocytes. In sharp contrast, natural killer (NK) cells are able to kill target cells in a HLA-independent way, i.e. these cells “sense” the absence or abnormal expression of HLA molecules to express their cytolytic capacities, provided that tumour cells display ligands for NK activating receptors.
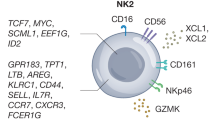
Morphologically, NK cells mostly appear as large granular lymphocytes. Cell surface phenotype defining human NK cell shows the absence of CD3 (excluding T cells) and the expression of CD56 and CD16. CD56 is the 140-kDa isoform of the neural cell adhesion molecule (NCAM) found on NK cells and a minority of T cells whose function is currently not defined. CD16 is the FcγRIIIa receptor responsible for antibody-dependent cell cytotoxicity (ADCC). The population of NK cells is phenotypically and functionally heterogeneous. The density of CD56 at NK cell surface discriminates between two functionally distinct NK cell subsets. The CD56bright NK subset represents 10% of circulating NK cells. This subset is characterized by a poor ability to kill tumour cell targets but produces high amount of cytokines. Conversely, the majority of circulating NK cells (CD56dim) has a high ability to spontaneously kill tumour cell targets but produces low amounts of cytokines. The existence of two functional NK cell subsets and the fact that CD56bright NK cells are present more in lymphoid organs support the notion that progression from CD56bright to CD56dim NK cells is likely part of a continuum in their development [1, 2]. NK cells do not express clonally distributed receptors for antigens but express receptors with opposite functions that finely regulate their activities. Physiologically, cells are protected from NK-mediated cytotoxicity by adequate expression of HLA class I molecules. Indeed, NK cells express at their surface HLA-specific inhibitory receptors [killer immunoglobulin-like receptors (KIR) and CD94/NKG2A/B heterodimers]. The recognition of normal HLA class I molecules on target cells downregulates the NK-mediated cytolytic activity [3]. Defects in HLA class I molecules expression and/or function occur in almost every type of solid tumour, although the frequency of these abnormalities varies markedly among the various types of malignancies [4]. HLA class I molecules expression has been investigated in B cell and Hodgkin lymphoma (HL), chronic lymphocytic leukaemia (CLL), acute lymphoblastic leukaemia (ALL) and acute myeloid leukaemia (AML), as summarized in Table 1. In the absence of these inhibitory signals, activating receptors, if engaged by ligands on the target cell surface, activate NK cytotoxicity. These concepts are the basis of the “missing self” hypothesis. Activating receptors transduce signals through their intracytoplasmic region containing the immunoreceptor tyrosine activating motif (ITAM) [5]. Natural cytotoxicity receptors (NCR) [6] and NKG2D [7] are the major receptors involved in NK cytotoxicity. Three NCR are expressed on NK cells: NKp30, NKp44 and NKp46. NKp30 and NKp46 are constitutively expressed, while NKp44 is only expressed on activated NK cells. Although the cellular ligands recognized by NCRs have not been fully characterized yet, these receptors were described to recognize viral proteins [8–10]. NKG2D is a C-type lectin-like receptor whose ligands, the MHC class I chain-related protein A (MICA) and B (MICB) and the family of UL16-binding proteins (ULBP), have recently been described [11]. The ligand–receptor pairs are described in Table 2.
Since in most haematological malignancies tumour cells are located in bone marrow (acute or chronic leukaemia, myeloma, myelodysplastic syndromes and sometimes lymphoma), NK role in the bone marrow environment is of interest. Nonetheless, few data are available regarding the distribution of NK cells in the bone marrow. Two old studies, involving a small number of healthy donors, have quantified the NK cells in the bone marrow with an average percentage of 6.3% CD16+ [12] or 4.4% of CD3−CD16+CD56+ marrow cells [13]. Recently, Freud et al. identified a novel CD34dim CD45RA+ hematopoietic precursor cell (HPC) that is integrin α4β7bright. This subset constitute less than 1% of BM CD34+ HPCs and about 6% of blood CD34+ HPCs, but more than 95% of lymph nodes (LN) CD34+ HPCs. After stimulation by IL-2, IL-15 or activated T cells, these CD34dim CD45RA+ α4β7bright become CD56bright NK cells. Thus, this unique subset of CD34+ HPCs could be produce in BM and traffics through the blood to the lymph nodes where it differentiates into CD56bright NK cells under the influence of endogenous cytokines [14]. This novel subset is interesting since some tumour cells, particularly lymphoma cells, invade LN. Unfortunately, no data are available regarding NK cells in BM or LN from patients with haematological malignancies, except personal data (A. Boehrer) showing decreased expression of activating receptors.
Several clues suggest that NK cells play an important role in the control and clearance of leukemic cells, the most impressive results having been obtained in allogeneic hematopoietic cell transplantation. In AML patients, HLA-C-mismatched transplantation has been shown to induce long-lasting remissions, which was attributed to the absence of appropriate KIR ligands [15]. In order to contribute to design immunotherapeutic approaches involving innate immunity in an autologous setting, we summarize in this review the various defects observed. We describe and classify these abnormalities observed in three categories: first, decreased or increased absolute number of NK cells; second, altered expression of receptors on NK cell surfaces, i.e. decrease of activating receptors or increase of inhibitory receptors; third, loss in functional cytotoxic abilities of NK cells. These data are summarized in Table 3 (description of the various studies) and in Table 4 (NK abnormalities observed in haematological malignancies). We then propose some clues for restoration of innate immunity functions in haematological malignancies.
Myeloid haemopathies
Myelodysplastic syndromes (MDS) are clonal hematopoietic stem cell disorders leading to peripheral cytopenias. MDS are classified into eight subtypes according to the WHO proposals. Several immunological abnormalities, such as hypo- or hyper-gammaglobulinemia, autoimmune diseases, peripheral lymphopenia, abnormal B or T cell function have been described in this clinical setting [16]. Reduced ADCC and decreased direct NK cell cytolytic functions have been reported in preleukemic syndromes more than 25 years ago [17]. In these early studies, decreased NK cells activity was not found to be related to a decreased absolute number of NK cells in peripheral blood or in bone marrow. However, Yokose et al. [18] reported later a decreased absolute number of CD3−CD16+ and CD3−CD56+ cell population in patients with high-risk MDS group (RAEB, t-RAEB and CMML), which was associated with an increased plasmatic level of sIL-2R. Recently, Epling-Burnette et al. [19] have shown that NKp30 expression is lower in MDS patients (40 ± 26% vs. 58 ± 17%) and that NKG2D expression is selectively decreased on NK cells from patients who exhibit low NK functions (62.5% of patients exhibiting an average of 6.1 ± 4.4% of specific lysis in a cytotoxic assay versus 40 ± 17% in healthy donors). However, decreased expression of NKp30 had not been found in a previous work by Kiladjian et al. [16]. Since NK cells phenotype could not explain the decreased lytic activity, Kiladjian et al. analysed expression of CD3ζ chain and seric soluble MICA (sMICA) concentration, which were also normal. They also showed that NK cells from MDS patients do not proliferate in vitro after IL-2 stimulation [16]. Elevated circulating levels of TNF could be an explanation for abnormal cytotoxicity [20].
The myeloproliferative syndromes (MPS) include chronic myelogenous leukaemia (CML), defined by the Philadelphia (Ph1) chromosome or its molecular equivalence BCR/ABL, and the Ph1-negative MPS, i.e. polycythemia vera (PV), essential thrombocythemia (ET) and idiopathic myelofibrosis (IMF). In the Ph1-negative classical MPS, an acquired activating mutation of the protein kinase JAK2 (JAK2 V617F) has recently been described. This mutation is present in nearly all PV patients, and in half patients with ET or IMF [21]. In 1989, Froom et al. [22] reported similar percentage of CD16+ cells in MPS patients and control. Some of these patients had a decreased NK activity in vitro, not corrected by stimulation with IL-2 or IFNα, suggesting that the NK cells defect could be intrinsic [22]. These conclusions have nonetheless been modified by more recent studies. Chang et al. [23] showed that functional NK cell deficiency in CML patients is restorable in vitro by IL-2. Pierson et al. [24] have described a progressive decrease in NK cell number and the loss of the effect of IL-2 as the disease progresses from chronic phase to blast crisis. Chiorean et al. [25] have shown that NK-92 cells transduced with the BCR/ABL oncogene proliferated indefinitely in the absence of IL-2 and exhibited decreased natural cytotoxicity against K562 targets but normal IL-2, IFNγ or TNFα production. Transduced NK-92 cells expressed CD158i, CD158j and CD158e receptors, suggesting that an imbalance in KIR expression, directly or indirectly linked to BCR/ABL expression, may modify NK cytotoxic function [25]. Imatinib mesylate, a BCR/ABL specific tyrosine kinase inhibitor used in frontline treatment of CML, inhibits the proliferation of BCR/ABL-transduced NK [25]. Boissel et al. [26] reported that CML patients had abnormally high seric levels of sMICA and weak NKG2D expression on NK cells. Imatinib mesylate therapy increases NKG2D expression and decreases MICA protein production and release, thus contributing to normal NK cytotoxicity through the restoration of a functional NKG2D signalling [26].
Gersuk et al. [27] have focused their attention on NK cells in Ph1-negative MPS. Percentage of NK cells (defined as CD16+ cells) is decreased in IMF and increased in PV patients. Furthermore, NK cytotoxic activity is decreased in Ph1-negative MPS patients, more severely in IMF patients [27]. Treatment of NK cells from patients with PV or ET (but not from patients with IMF) by IFNα or IL-2 increases their cytotoxicity against K562 [27]. In order to analyse more precisely NK population in PV, we used the more recent definition for NK cells, i.e. CD3− CD16+ CD56+ cells instead of the CD16+ gate used by Gersuk et al. [27]. We have shown that the percentage and absolute number of NK cells are significantly increased in PV, but we failed to detect any abnormalities in the expression of activating NK cell receptors, while, in line with Gersuk’s results, we observed decreased NK degranulation capacities. Furthermore, we showed an increased expression of KIR2DL1 (CD158a), an inhibitory receptor, on NK cells from PV patients (personal observation, C. Sanchez). Together, these data suggest that NK cytotoxic defect may be related to impaired signalling despite normal expression of receptors. A potential link between impaired NK functions and IMF is provided by PDGF, since its concentration is significantly elevated in IMF patients and inhibits NK activity.
Regarding acute myeloid leukaemia (AML), in addition to data from KIR mismatch in allogeneic stem cell transplantation [15], several clues support an antileukaemia role for autologous NK cells in AML. Impaired NK cell function and cytokine production are associated with early relapse in AML. The poor cytotoxicity of NK cells from AML patients could be explained by insufficient NCR/ligand interactions [28]. Sivori et al. [29] have described, in healthy individuals, NK cells characterized by low expression of NCR activating receptors, referred as NCRdull phenotype. This NCRdull phenotype is rare in healthy individuals (<10%), while it is found in the majority of patients with AML [28]. Since one of the NCR, i.e. NKp46, has a pivotal role regarding the ability of human NK cells to kill tumour targets [30], its low expression could explain the defective NK cytotoxicity against leukaemia cells observed in AML patients [28]. This defect in NCR expression could be potentiated by the fact that leukemic cells express low amounts of both NCR and NKG2D ligands [31]. To further support the antileukaemia role of NK cells in AML, Fauriat et al. [32] have shown a significant increase of the expression of the NKp30 and NKp46 after complete remission in AML patients, which was paralleled by the restoration of NCR-driven cytotoxicity. Fauriat et al. [32] also showed that the NCRdull phenotype was induced by leukemic cells coculture with either developing or mature NK cells, and required cell-to-cell contact between leukaemia and NK cells. Another way to escape from NK surveillance has been described by Salih et al. [33]: most AML patients had elevated levels of sMICA and sMICB, that could lead to ligand shedding and trigger internalisation of surface NKG2D, thus impairing NK cell function.
Myeloma and lymphoid haemopathies
Multiple myeloma (MM), despite initial response to conventional treatments and promising results with novel agents, is the paradigm of an incurable malignancy [34]. In MM, normal numbers of NK cells are present in peripheral blood [35], but NK cytotoxicity is altered [36, 37]. Expression of NCR and NKG2D is normal but the expression of CD16 and 2B4/CD244, an activating coreceptor, is decreased [38]. In addition to deficient NK function, plasma cells display variable susceptibility to NK control. Carbone et al. [39] have shown that normal NK efficiently kill early-stage MM cells by a NCR and NKG2D-dependent pathway. In contrast, late-stage MM cells are protected from NK lysis by high expression of HLA class I molecules. The expression of NCAM was analyzed but conclusions on its beneficial or deleterious effect are controversial [40]. Constitutive expression of MICA on human tumours promotes ligand shedding, which triggers internalisation of surface NKG2D and thus impairs NK cell function [41]. Although monoclonal gammopathy of undetermined significance (MGUS) and MM are considered to be the same entity, significant differences are observed regarding MICA expression. Plasma cells from MM patients have low MICA surface expression and significant levels of circulating sMICA while plasma cells from MGUS patients exhibit opposite characteristics [42]. Interestingly, MGUS patients frequently develop anti-MICA antibodies that antagonize the suppressive effects of sMICA [42]. This may constitute an interesting, although not totally efficient, attempt to self-restoration of the anti-tumour immune response. Another mechanism for plasma cells to escape from NK cell killing could be the expression of HLA-G, an HLA class I molecule which is able to inhibit the effector functions of both cytotoxic T lymphocytes and NK cells via the engagement of two receptors, ILT2/CD85j and KIR2DL4 [43]. Multiple HLA-G transcripts are detected by RT-PCR but HLA-G cell surface expression is low or undetectable on MM cells [37]. These results suggest post-transcriptional regulatory mechanisms and/or the expression of soluble isoforms that have immunosuppressive and pro-apoptotic effects on NK [44].
In chronic lymphocytic leukaemia (CLL), the total number of NK cells in the peripheral blood is increased ([45]; personal observations, B. Knoblauch), but these NK cells have defective cytotoxic activity [37]. This cytotoxic activity can be stimulated in the presence of recombinant human IFNα or IL-2 [46]. NK cells from CLL patients have a normal tumour-cell binding capacity but fail to release sufficient amounts of soluble cytolytic molecules upon activation. While the expression of one or more HLA class I alleles on leukemic cells is downregulated in 85% of CLL samples [47], leukemic cells do not express either MICA nor ULBPs (except a low expression of ULBP3) [48]. Thus, in the absence of NKG2D engagement by their ligands at the surface of CLL cells, the defect in HLA class I molecules expression could be insufficient to trigger effective NK cytotoxicity. Finally, a variable expression of HLA-G at tumour cell surface has been observed in CLL, and could contribute to tumour escape from NK lysis via the engagement of its ligand that negatively regulates NK cytotoxicity [37].
In acute lymphoblastic leukaemia (ALL), in contrast with AML, KIR mismatch does not improve the disease-free survival, suggesting that ALL cells exhibit, in addition to KIRs protection, additional mechanisms of resistance to NK lysis [49]. Several studies have demonstrated a reduced NK cell activity in patients with ALL [50]. Reid et al. [51] have shown that the killing of the pre-B ALL cell lines does not correlate with the lymphocyte function-associated antigen-1 (LFA-1) and the intercellular adhesion molecule-1 (ICAM-1) expression, although the LFA-1/ICAM-1 interaction is the dominant adhesion pathway for NK cells. Romanski et al. [49] showed selective resistance of B-precursor ALL to cytotoxicity mediated by the NK-92 activated NK cell line, that could be explained by the interaction between HLA-G and the inhibitory receptor KIR2DL4. Nevertheless, this mechanism seems unlikely in pre-B ALL, thus suggesting that defective engagement of activating receptors, rather than activation of inhibitory receptors, is responsible for ALL resistance to NK lysis. In line with this hypothesis, expression of the NKG2D activating receptor ligands MICA/B was only observed in NK-sensitive T-ALL cell line, while NK-resistant B-ALLs did not express detectable amounts of MICA/B [49]. Deficient engagement of other activating receptors may also contribute to ALL resistance to NK lysis, since Pende et al. [52] have reported that B-ALL cells lose or express low levels of several other NK activating ligands such as ULBPs, PVR, Nectin-2, CD48 or NK-T-B antigen.
Potential use of NK cells in active immunotherapy
This review has pointed out important NK cells abnormalities in virtually all studied haematological malignancies. Attempts to cure such malignancies rely on chemotherapy and immunotherapy. Specific immunotherapy has many limitations, mainly abnormal expression of HLA class I molecules. This has raised some hope in the concept of innate immunity modulation, particularly involving NK cells. For the sake of clarity, the therapeutic proposals can be divided in two categories: therapeutic options aiming to enhance NK cell cytotoxicity and/or target cell susceptibility, and novel therapeutic tools specifically designed to target tumour cells, and which, in addition, have shown immunomodulatory properties towards NK cells.
Specific NK approaches As previously stated, KIR mismatch in allogeneic transplantation is not the scope of this review, and thus will not be discussed. Several studies have investigated NK cell reconstitution in patients undergoing haplotype-matched (10 patients) [53] or haplotype-mismatched (8 patients) [54] stem cell transplantation (SCT) as a treatment of AML or accelerated phase of CML. NK cells recovered within the first month after transplantation and rapidly reached normal counts [55]. In contrast, T cells reconstituted later and were usually not detectable until the fourth month after transplantation. Nevertheless, this NK cell reconstitution is abnormal with an increase of the CD56bright subset, especially during the first 3 months, which could explain the decrease in NK cytotoxicity [53, 54]. Patterns of NK cell receptors expression during NK reconstitution were also analysed. Regarding the expression of NCR, the results vary from study to study. NKp30 levels appear to be decreased after SCT and then restored between 1 and 4–6 months after [53, 56]. Data for NKp46 are controversial: Vitale et al. described decrease in its expression while Schulze et al. reported its upregulation [54, 56]. In this last study, NKp46 upregulation was associated to a reduced expression of NKG2D. Nguyen et al. [53] also demonstrated that the frequency of KIR-positive NK cells remained very low for all patients and that the frequency of CD94/NKG2A and KIR were inversely correlated. Low expression of NKp30 and predominance of CD56bright NK cells are arguments in favour of the persistence of immature NK cells.
Although the anti-tumour role of NK is pivotal in allogenic transplantation, this therapeutic option is not available for all patients, suggesting the need for other possibilities of NK-induced anti-tumour response. The use of genetically modified NK cells can redirect, improve or induce de novo their recognition and killing of tumour cells. Such innovative approaches have been developed by the transduction of NK cells with a chimeric receptor directed at the B lymphocyte antigens CD19 and CD20, or against the ERBB2 oncogene [57–59]. A more traditional approach consists in the development of monoclonal antibodies (mAbs) directed against the haematological target cell. For example, de Romeuf et al. [60] generated a chimeric anti-CD20 which displayed improved FcγRIIIa/CD16 binding and FcγRIIIa-dependent effector function. Since NK cell activity is controlled by a balance between inhibitory and activating receptors, using blocking mAbs raised against NK cell inhibitory receptors should theoretically enhance the anti-tumour response of NK cells [61]. Nevertheless, the possible pitfall of such a therapy is the breakdown of tolerance to self HLA, resulting in the development of autoimmune diseases. Another possibility is to genetically link cytokine to mAbs in order to improve their efficiency, since this method would allow the induction of NK cells and T cells proliferation at the tumour site [62].
Infusion of NK cells with increased cytotoxic activity is another emerging tool for cancer therapy. This can be achieved by culture with cytokines such as IFN type I, IL-2, IL-12, IL-15, IL-18 or IL-21 [62–64]. IL-2 has been widely studied since this cytokine activates and expands NK cells, but its toxic effects limit its clinical use. From an immunological point of view, the use of IL-2 is limited by its capacity to trigger Treg cells, which inhibit NK cells through the downregulation of NKp30 and NKG2D by TGF-β. Nevertheless, this limitation could be overcome, since it has been recently demonstrated that Treg depletion resulted in an improved survival in a leukaemia murine model [65]. IL-21 is another cytokine that looks particularly promising in therapeutics since it induces both the proliferation and the cytotoxicity of the cytokine-secreting CD56bright NK subset [66], while downregulating the suppressive effects of Treg cells. IL-15 is also of potential interest in the modulation of NK cells activity. Two experiments in mice showed the effects of IL-15 on NK cells: mice IL15−/− or IL-15Rα−/− have reduced numbers of NK (and memory CD8+ T) cells when mice constitutively expressing IL-15 display an increased resistance to tumour growth [67]. IL-15 activates not only NK cells but also T lymphocytes, inhibits apoptosis of activated T cells and maintains the CD8+ memory T cell subpopulation [67].
One another interesting approach consists in NK stimulation via dendritic cells (DCs). DCs release IL-12, IL-15 and IL-18 that participate in NK cell proliferation, activation and survival. In response, NK cells produce IFNγ and TNFα. This continual release of cytokines results in a positive feedback loop between NK and DC. Activation of DC can be obtained in multiple ways, among which the stimulation via Toll-like receptors (TLRs), particularly TLR9, has raised interest. The action of TLR9 agonists on plasmacytoid DC induces the secretion of type I IFN that activates NK cells, but also inhibits the generation of Treg cells, thus may further contribute to the anti-tumour potential of NK cells [68].
Another alternative is the use of chemokines, which are molecules involved in chemotaxis of immune cells, including their recruitment to the site of inflammation or activation. The chemokines produced and regulating NK cell functions are outlined in Table 5. A role for CXCR3 and CXCR4, as well as for its ligand CXCL12, has been described in NK cell homing to the BM whereas CCR7, CCL19 and CCL21 seems to be involved in LN homing [69]. Some chemokines are overexpressed in haematological malignancies: CXCR4 in CLL and AML, CCR7 in T-ALL, CCL22 in T, B and Hodgkin’s lymphoma [70]. Furthermore, in AML, three release clusters have been identified: CCL2-4/CXCL1/8, CCL5/CXCL9-11 and CCL13/17/22/24/CXCL5. These three clusters differ in their T cell chemotaxis towards the leukemic cells [71]. Nonetheless, a possible pitfall is that NK cells may damage the affected tissues, if an abnormal HLA expression is present [72].
Finally, the use of association of post-consolidation immunotherapy with histamine dihydrochloride (HDC) and IL-2 has been shown to reduce the risk of relapse in AML with a 3-year leukaemia-free survival for patients treated with HDC and IL-2 of 40 versus 26% in the control arm [73]. Interestingly, one probable explanation for such a favourable effect is the protection of NK cell from the downregulation of activating receptor expression induced by leukemic cells [74].
Recently, novel drugs used in haematology raised a great interest regarding their NK cell immunomodulatory properties. The efficacy of old anticancer therapeutic approaches has recently been related to their effects on NK cells. The best example is the so-called “BCG therapy” in bladder cancer, the efficiency of which relies, at least in part, on the influx of NK cells at tumour site [75]. Recently many novel drugs have emerged in the management of haematological malignancies: tyrosine kinase inhibitors (TKI), immunomodulating drugs (IMIDs®), proteasome inhibitors (bortezomib), demethylating agents (such as azacytidine) or inhibitors of histone deacetylase (HDACis). The mechanisms of action of these drugs are not always fully understood, suggesting additional effects to the direct cytotoxicity against tumours, like enhancing NK cytotoxicity or increase in target cell susceptibility to NK lysis.
TKI affect BCR/ABL but also other kinases like the stem cell factor receptor (c-kit) or platelet-derived growth factor receptor (PDGFR) which are involved in the activation of immune effector cells. Regarding the target cell, Salih et al. [33] focused their study on the effects of imatinib, dasatinib and nilotinib on the myeloid leukaemia cell line K562 cell, that harbour the BCR/ABL mutation. Expression of NKG2D ligands on K562 cells exposed to pharmacological concentrations of one of the three TKI was diminished to a similar extent. This resulted in reduced NK cell cytotoxicity against K562 cell line, and diminished IFNγ production by NK cells. Regarding effector cells, the three TKI have different effects. Nilotinib inhibits cytokine production by NK, probably by preferential induction of cell death in the CD56bright NK subset. In contrast, dasatinib interacts with NK cells at an early stage of signal transduction, preventing PI3K phosphorylation, so that the effects observed in response to dasatinib could possibly be a consequence of impaired target cell recognition and not directly of diminished cytotoxic function. Finally, effects of imatinib on NK cells is probably indirect since imatinib fosters DC/NK reciprocal activation, resulting in the enhancement of NK cells anti-tumoural function [76].
The IMIDs® (thalidomide, lenalidomide) are currently used in the treatment of MM or MDS and tested in other haematological malignancies such as CLL or non-Hodgkin lymphoma. IMIDs® have immunoregulatory, anti-tumour and anti-angiogenic properties but Reddy et al. [77] suggested that the major mechanism of action of IMIDs® is activation of the innate immune system and more particularly NK cells. In a murine model, exposure to IMIDs® lead to the recruitment of NK cells via stimulation of DCs and modification of the cytokine microenvironment associated with an increase in monocyte chemotactic protein-1 (MCP-1), TNFα, IFNγ and probably augmented ADCC by triggering IL-2 secretion by T lymphocytes.
Bortezomib, a proteasome inhibitor, is used in treatment of MM. Bortezomib sensitizes tumours cells to chemotherapeutic drugs or radiations by upregulation of DR5, a TRAIL ligand expressed on NK cells [78]. Other studies reported a downregulation of HLA class I molecules [79] or an upregulation of DNAM-A and NKG2D ligands [80] in MM plasmocytes after bortezomib regimen, thus favouring NK–plasma cell interaction and destruction. Butler et al. [81] observed that bortezomib, and other proteasome inhibitor drugs with distinct mechanisms of action, highly and specifically upregulated ULBP1 mRNA and cell surface protein in head and neck squamous cell carcinoma cells. Although these reports suggest an activating effect of bortezomib on immune cell functions, in contrast Wang et al. [82] reported that the proteasome inhibition induces apoptosis in primary human NK cells and suppresses NKp46-mediated cytotoxicity.
Demethylating agents such as 5-azacytidine or 5-aza-2′-deoxycytidine are used in treatment of MDS, since DNA hypermethylation, notably of tumour-suppressor genes, is supposed to have a pivotal role in leukaemogenesis. The effects of these drugs are complex. Two studies have shown that 5-aza-2′-deoxycytidine, used alone or in combination with other drugs, upregulates the expression of the NKG2D ligands, ULBP and MICB, increasing the susceptibility of tumour cells to NK cells. This mechanism could be related to promoter DNA methylation [83, 84]. A pitfall in this approach is that KIRs expression is also controlled by DNA methylation, so that demethylating drugs could induce KIRs transcription and finally decrease NK cytotoxic activity [85].
HDACis, like vorinostat or panobinostat, are recently developed drugs targeting the epigenetic regulation of cancer. Their anti-tumour effect involves triggering of differentiation, cycle-cell arrest and apoptosis of tumour cells [86]. Vorinostat, valproic acid, sodium butyrate and trichostatin A upregulate the expression of NKG2D ligands (MICA and MICB) and DNAM-1 on leukemic cells, thus improving NK-mediated killing via NKG2D [87–89]. Furthermore, HDACis could suppress NK cytotoxicity via impaired granule exocytosis and downregulation of NKp30 and NKp46, which could be a consequence of NFκB inhibition [90]. Sodium butyrate can also stimulate the expression of Sp1 and the binding of both Sp1 and heat shock transcription factor 1 (HSF1) to the MICA/B promoter, finally leading to an increased production of these NKG2D ligands [88].
Conclusion
The discovery of HLA class I-specific NK receptors, together with studies on NK cell function and regulation, molecular properties and genetics, provides an interesting possibility for innovative immunotherapy approach of haematological malignancies. Several mechanisms are involved in the alterations of NK cell functions described in haematological malignancies: decreased expression of activating receptors, increased expression of inhibitory receptors, defective expression of ligands on target cells or defective cytotoxic armamentarium. These abnormalities provide some clues for immunotherapy protocols using monoclonal antibodies, novel drugs or cytokines. Recently, many novel drugs used against haematological malignancies have shown immunomodulatory properties on NK cells, instead of the potent and wide (B, T and NK lymphocytes) immunosuppressive effects of classical and high-dose chemotherapy. These drugs offer an opportunity to simultaneously attack tumour cells directly and putatively enhance the immune response. This might provide a less toxic and long-lasting (via immune modulation) anti-tumour effect hopefully protecting from relapse in haematological malignancies.
References
Caligiuri MA (2008) Human natural killer cells. Blood 112:461–469
Vivier E (2006) What is natural in natural killer cells? Immunol Lett 107:1–7
Bottino C, Moretta L, Pende D et al (2004) Learning how to discriminate between friends and enemies, a lesson from natural killer cells. Mol Immunol 41:569–575
Garrido F, Cabrera T, Lopez-Nevot MA et al (1995) HLA class I antigens in human tumors. Adv Cancer Res 67:155–195
Biassoni R, Cantoni C, Pende D et al (2001) Human natural killer cell receptors and co-receptors. Immunol Rev 181:203–214
Moretta A, Biassoni R, Bottino C et al (2000) Natural cytotoxicity receptors that trigger human NK-cell-mediated cytolysis. Immunol Today 21:228–234
Watzl C (2003) The NKG2D receptor and its ligands—recognition beyond the “missing self”? Microbes Infect 5:31–37
Arnon TI, Achdout H, Levi O et al (2005) Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol 6:515–523
Arnon TI, Lev M, Katz G et al (2001) Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol 31:2680–2689
Mandelboim O, Lieberman N, Lev M et al (2001) Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409:1055–1060
Biassoni R, Cantoni C, Marras D et al (2003) Human natural killer cell receptors: insights into their molecular function and structure. J Cell Mol Med 7:376–387
Clark P, Normansell DE, Innes DJ et al (1986) Lymphocyte subsets in normal bone marrow. Blood 67:1600–1606
Rego EM, Garcia AB, Viana SR et al (1998) Age-related changes of lymphocyte subsets in normal bone marrow biopsies. Cytometry 34:22–29
Freud AG, Becknell B, Roychowdhury S et al (2005) A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity 22:295–304
Ruggeri L, Capanni M, Urbani E et al (2002) Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 295:2097–2100
Kiladjian JJ, Bourgeois E, Lobe I et al (2006) Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 20:463–470
Kerndrup G, Meyer K, Ellegaard J et al (1984) Natural killer (NK)-cell activity and antibody-dependent cellular cytotoxicity (ADCC) in primary preleukemic syndrome. Leuk Res 8:239–247
Yokose N, Ogata K, Ito T et al (1994) Elevated plasma soluble interleukin 2 receptor level correlates with defective natural killer and CD8+ T-cells in myelodysplastic syndromes. Leuk Res 18:777–782
Epling-Burnette PK, Bai F, Painter JS et al (2007) Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 109:4816–4824
Verhoef GE, De Shouwer P, Ceuppens JL et al (1992) Measurement of serum cytokine levels in patients with myelodysplastic syndromes. Leukemia 6:1268–1272
James C, Ugo V, Le Couedic JP et al (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434:1144–1148
Froom P, Aghai E, Kinarty A et al (1989) Decreased natural killer (NK) activity in patients with myeloproliferative disorders. Cancer 64:1038–1040
Chang WC, Fujimiya Y, Casteel N et al (1989) Natural killer cell immunodeficiency in patients with chronic myelogenous leukemia. III. Defective interleukin-2 production by T-helper and natural killer cells. Int J Cancer 43:591–597
Pierson BA, Miller JS (1996) CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood 88:2279–2287
Chiorean EG, Dylla SJ, Olsen K et al (2003) BCR/ABL alters the function of NK cells and the acquisition of killer immunoglobulin-like receptors (KIRs). Blood 101:3527–3533
Boissel N, Rea D, Tieng V et al (2006) BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol 176:5108–5116
Gersuk GM, Carmel R, Pattamakom S et al (1993) Quantitative and functional studies of impaired natural killer (NK) cells in patients with myelofibrosis, essential thrombocythemia, and polycythemia vera. I. A potential role for platelet-derived growth factor in defective NK cytotoxicity. Nat Immun 12:136–151
Costello RT, Sivori S, Marcenaro E et al (2002) Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 99:3661–3667
Sivori S, Pende D, Bottino C et al (1999) NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol 29:1656–1666
Moretta A, Bottino C, Vitale M et al (2001) Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol 19:197–223
Pende D, Cantoni C, Rivera P et al (2001) Role of NKG2D in tumor cell lysis mediated by human NK cells: cooperation with natural cytotoxicity receptors and capability of recognizing tumors of nonepithelial origin. Eur J Immunol 31:1076–1086
Fauriat C, Just-Landi S, Mallet F et al (2007) Deficient expression of NCR in NK cells from acute myeloid leukemia: evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 109:323–330
Salih HR, Antropius H, Gieseke F et al (2003) Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 102:1389–1396
Pant S, Copelan EA (2007) Hematopoietic stem cell transplantation in multiple myeloma. Biol Blood Marrow Transplant 13:877–885
King MA, Radicchi-Mastroianni MA (1996) Natural killer cells and CD56+ T cells in the blood of multiple myeloma patients: analysis by 4-colour flow cytometry. Cytometry 26:121–124
Matsuzaki H, Kagimoto T, Oda T et al (1985) Natural killer activity and antibody-dependent cell-mediated cytotoxicity in multiple myeloma. Jpn J Clin Oncol 15:611–617
Maki G, Hayes GM, Naji A et al (2008) NK resistance of tumor cells from multiple myeloma and chronic lymphocytic leukemia patients: implication of HLA-G. Leukemia 22:998–1006
Fauriat C, Mallet F, Olive D, Costello RT (2006) Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia 20:732–733
Carbone E, Neri P, Mesuraca M et al (2005) HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 105:251–258
Jarahian M, Watzl C, Issa Y et al (2007) Blockade of natural killer cell-mediated lysis by NCAM140 expressed on tumor cells. Int J Cancer 120:2625–2634
Groh V, Wu J, Yee C et al (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419:734–738
Jinushi M, Vanneman M, Munshi NC et al (2008) MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA 105:1285–1290
Saverino D, Fabbi M, Ghiotto F et al (2000) The CD85/LIR-1/ILT2 inhibitory receptor is expressed by all human T lymphocytes and down-regulates their functions. J Immunol 165:3742–3755
Rouas-Freiss N, Moreau P, Ferrone S et al (2005) HLA-G proteins in cancer: do they provide tumor cells with an escape mechanism? Cancer Res 65:10139–10144
Palmer S, Hanson CA, Zent CS et al (2008) Prognostic importance of T and NK-cells in a consecutive series of newly diagnosed patients with chronic lymphocytic leukaemia. Br J Haematol 141:607–614
Katrinakis G, Kyriakou D, Papadaki H et al (1996) Defective natural killer cell activity in B-cell chronic lymphocytic leukaemia is associated with impaired release of natural killer cytotoxic factor(s) but not of tumour necrosis factor-alpha. Acta Haematol 96:16–23
Verheyden S, Ferrone S, Mulder A et al (2009) Role of the inhibitory KIR ligand HLA-Bw4 and HLA-C expression levels in the recognition of leukemic cells by natural killer cells. Cancer Immunol Immunother 58:855–865
Poggi A, Venturino C, Catellani S et al (2004) Vdelta1 T lymphocytes from B-CLL patients recognize ULBP3 expressed on leukemic B cells and up-regulated by trans-retinoic acid. Cancer Res 64:9172–9179
Romanski A, Bug G, Becker S et al (2005) Mechanisms of resistance to natural killer cell-mediated cytotoxicity in acute lymphoblastic leukemia. Exp Hematol 33:344–352
Sorskaar D, Lie SO, Forre O (1985) Natural killer cell activity of peripheral blood and bone marrow mononuclear cells from patients with childhood acute lymphoblastic leukemia. Acta Paediatr Scand 74:433–437
Reid GS, Bharya S, Klingemann HG et al (2002) Differential killing of pre-B acute lymphoblastic leukaemia cells by activated NK cells and the NK-92 ci cell line. Clin Exp Immunol 129:265–271
Pende D, Castriconi R, Romagnani P et al (2006) Expression of the DNAM-1 ligands, Nectin-2 (CD112) and poliovirus receptor (CD155), on dendritic cells: relevance for natural killer-dendritic cell interaction. Blood 107:2030–2036
Nguyen S, Dhedin N, Vernant JP et al (2005) NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 105:4135–4142
Schulze A, Schirutschke H, Oelschlagel U et al (2008) Altered phenotype of natural killer cell subsets after haploidentical stem cell transplantation. Exp Hematol 36:378–389
Buser A, Stern M, Arber C et al (2008) Impaired B-cell reconstitution in lymphoma patients undergoing allogeneic HSCT: an effect of pretreatment with rituximab? Bone Marrow Transplant 42:483–487
Vitale C, Chiossone L, Morreale G et al (2004) Analysis of the activating receptors and cytolytic function of human natural killer cells undergoing in vivo differentiation after allogeneic bone marrow transplantation. Eur J Immunol 34:455–460
Imai C, Iwamoto S, Campana D (2005) Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 106:376–383
Muller T, Uherek C, Maki G et al (2008) Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother 57:411–423
Pegram HJ, Jackson JT, Smyth MJ et al (2008) Adoptive transfer of gene-modified primary NK cells can specifically inhibit tumor progression in vivo. J Immunol 181:3449–3455
de Romeuf C, Dutertre CA, Le Garff-Tavernier M et al (2008) Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br J Haematol 140:635–643
Sheridan C (2006) First-in-class cancer therapeutic to stimulate natural killer cells. Nat Biotechnol 24:597
Waldmann TA (2006) The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol 6:595–601
Xiang J, Chen Z, Huang H et al (2001) Regression of engineered myeloma cells secreting interferon-gamma-inducing factor is mediated by both CD4(+)/CD8(+) T and natural killer cells. Leuk Res 25:909–915
Chan CJ, Andrews DM, Smyth MJ (2008) Can NK cells be a therapeutic target in human cancer? Eur J Immunol 38:2964–2968
Hallett WH, Ames E, Alvarez M et al (2008) Combination therapy using IL-2 and anti-CD25 results in augmented natural killer cell-mediated antitumor responses. Biol Blood Marrow Transplant 14:1088–1099
Wendt K, Wilk E, Buyny S et al (2007) Interleukin-21 differentially affects human natural killer cell subsets. Immunology 122:486–495
Kobayashi H, Dubois S, Sato N et al (2005) Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood 105:721–727
Krieg AM (2007) Development of TLR9 agonists for cancer therapy. J Clin Invest 117:1184–1194
Gregoire C, Chasson L, Luci C et al (2007) The trafficking of natural killer cells. Immunol Rev 220:169–182
Catusse J, Wollner S, Leick M et al (2010) Attenuation of CXCR4 responses by CCL18 in acute lymphocytic leukemia B cells. J Cell Physiol [Epub ahead of print]
Kittang AO, Hatfield K, Sand K et al (2010) The chemokine network in acute myelogenous leukemia: molecular mechanisms involved in leukemogenesis and therapeutic implications. Curr Top Microbiol Immunol [Epub ahead of print]
Maghazachi AA (2010) Role of chemokines in the biology of natural killer cells. Curr Top Microbiol Immunol [Epub ahead of print]
Brune M, Castaigne S, Catalano J et al (2006) Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: results of a randomized phase 3 trial. Blood 108:88–96
Romero AI, Thoren FB, Aurelius J et al (2009) Post-consolidation immunotherapy with histamine dihydrochloride and interleukin-2 in AML. Scand J Immunol 70:194–205
Brandau S, Riemensberger J, Jacobsen M et al (2001) NK cells are essential for effective BCG immunotherapy. Int J Cancer 92:697–702
Borg C, Terme M, Taieb J et al (2004) Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest 114:379–388
Reddy N, Hernandez-Ilizaliturri FJ, Deeb G et al (2008) Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol 140:36–45
Lundqvist A, Abrams SI, Schrump DS et al (2006) Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res 66:7317–7325
Shi J, Tricot GJ, Garg TK et al (2008) Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood 111:1309–1317
Soriani A, Zingoni A, Cerboni C et al (2009) ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113:3503–3511
Butler JE, Moore MB, Presnell SR et al (2009) Proteasome regulation of ULBP1 transcription. J Immunol 182:6600–6609
Wang X, Ottosson A, Ji C et al (2009) Proteasome inhibition induces apoptosis in primary human natural killer cells and suppresses NKp46-mediated cytotoxicity. Haematologica 94:470–478
Rohner A, Langenkamp U, Siegler U et al (2007) Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leuk Res 31:1393–1402
Tang KF, He CX, Zeng GL et al (2008) Induction of MHC class I-related chain B (MICB) by 5-aza-2′-deoxycytidine. Biochem Biophys Res Commun 370:578–583
Santourlidis S, Trompeter HI, Weinhold S et al (2002) Crucial role of DNA methylation in determination of clonally distributed killer cell Ig-like receptor expression patterns in NK cells. J Immunol 169:4253–4261
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358:1148–1159
Kato N, Tanaka J, Sugita J et al (2007) Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia 21:2103–2108
Zhang C, Wang Y, Zhou Z et al (2009) Sodium butyrate upregulates expression of NKG2D ligand MICA/B in HeLa and HepG2 cell lines and increases their susceptibility to NK lysis. Cancer Immunol Immunother 58:1275–1285
Diermayr S, Himmelreich H, Durovic B et al (2008) NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood 111:1428–1436
Ogbomo H, Michaelis M, Kreuter J et al (2007) Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett 581:1317–1322
Riemersma SA, Jordanova ES, Schop RF et al (2000) Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood 96:3569–3577
Murray PG, Constandinou CM, Crocker J et al (1998) Analysis of major histocompatibility complex class I, TAP expression, and LMP2 epitope sequence in Epstein–Barr virus-positive Hodgkin’s disease. Blood 92:2477–2483
Nouri AM, Smith S, Oliver TR et al (1998) Comparative expression of major histocompatibility complex (MHC) antigens on CD5+ and CD5− B cells in patients with chronic lymphocytic leukaemia (CLL). Eur J Cancer 34:1618–1622
Brouwer RE, van der Heiden P, Schreuder GM et al (2002) Loss or downregulation of HLA class I expression at the allelic level in acute leukemia is infrequent but functionally relevant, and can be restored by interferon. Hum Immunol 63:200–210
Masuda K, Hiraki A, Fujii N et al (2007) Loss or down-regulation of HLA class I expression at the allelic level in freshly isolated leukemic blasts. Cancer Sci 98:102–108
Wetzler M, Baer MR, Stewart SJ et al (2001) HLA class I antigen cell surface expression is preserved on acute myeloid leukemia blasts at diagnosis and at relapse. Leukemia 15:128–133
Demanet C, Mulder A, Deneys V et al (2004) Down-regulation of HLA-A and HLA-Bw6, but not HLA-Bw4, allospecificities in leukemic cells: an escape mechanism from CTL and NK attack? Blood 103:3122–3130
Vollmer M, Li L, Schmitt A et al (2003) Expression of human leucocyte antigens and co-stimulatory molecules on blasts of patients with acute myeloid leukaemia. Br J Haematol 120:1000–1008
Lemoli RM, Salvestrini V, Bianchi E et al (2009) Molecular and functional analysis of the stem cell compartment of chronic myelogenous leukemia reveals the presence of a. Blood 114:5191–5200
Verheyden S, Bernier M, Demanet C (2004) Identification of natural killer cell receptor phenotypes associated with leukemia. Leukemia 18:2002–2007
Sconocchia G, Lau M, Provenzano M et al (2005) The antileukemia effect of HLA-matched NK and NK-T cells in chronic myelogenous leukemia involves NKG2D-target-cell interactions. Blood 106:3666–3672
Robertson MJ (2002) Role of chemokines in the biology of natural killer cells. J Leukoc Biol 71:173–183
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sanchez, C.J., Le Treut, T., Boehrer, A. et al. Natural killer cells and malignant haemopathies: a model for the interaction of cancer with innate immunity. Cancer Immunol Immunother 60, 1–13 (2011). https://doi.org/10.1007/s00262-010-0898-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-010-0898-x