
Abstract
Despite being of the myeloid lineage, acute myeloid leukaemia (AML) blasts are of low immunogenicity, probably because they lack the costimulatory molecule CD80 and secrete immunosuppressive factors. We have previously shown that in vitro stimulation of autologous peripheral blood mononuclear cells (PBMCs) with primary AML cells modified to express CD80 and IL-2 promotes proliferation, secretion of Th1 cytokines and expansion of activated CD8+ T cells. In this study, we show that allogeneic effector cells (from a healthy donor or AML patients) when stimulated with IL-2/CD80 modified AML blasts were able to induce the lysis of unmodified AML blasts. Effector cells stimulated with IL-2/CD80AML blasts had higher lytic activity than cells stimulated with AML cells expressing CD80 or IL-2 alone. Similarly, AML patient PBMCs primed with autologous IL-2/CD80 AML cells had a higher frequency of IFN-γ secreting cells and show cytotoxicity against autologous, unmodified blasts. Crucially, the response appears to be leukaemia specific, since stimulated patient PBMCs show higher frequencies of IFN-γ secreting effector cells in response to AML blasts than to remission bone marrow cells from the same patients. Although studied in a small number of heterogeneous patient samples, the data are encouraging and support the continuing development of vaccination for poor prognosis AML patients with autologous cells genetically modified to express IL-2/CD80.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukaemia (AML) is a group of clonal haematopoietic stem cell disorders in which over-proliferation and failure to differentiate result in an accumulation of myeloblasts in the bone marrow. Despite advances in intensive chemotherapy and haematopoietic stem cell transplantation (HSCT), more than half of adult AML patients still die of their disease [1, 2]. One area which holds promise for improved treatment is immunotherapy (for recent reviews see [3, 4]). HSCT and donor leucocyte infusions (DLI) have been shown to confer clinically beneficial immune responses [5] and a number of putative leukaemia-associated antigens have recently been identified [6–10]. However, leukaemia patients often show disease-related [11–14] or treatment-induced [15–17] immunosuppression, making the challenge for immunotherapy considerable.
We are pursuing a whole cell vaccination strategy utilising genetically modified intact primary AML cells. This has the potential advantage of stimulating responses to multiple antigens and does not require knowledge of T cell epitopes or tailoring to individual patient HLA composition. Unlike many other malignancies, it is relatively easy to obtain large numbers of AML cells, and they are generally viable in vitro and recover well from frozen. AML cells should provide a unique opportunity for whole cell vaccination, as unlike many other tumour cells they express MHC II and other important immune-activating molecules such as ICAM1 and CD86 [18]. However, they are of low immunogenicity, probably because they lack expression of CD80 [18], express high levels of CLIP [19] and secrete immunosuppressive factors [20, 21]. We have therefore genetically modified primary AML cells to express CD80, either alone or in combination with IL-2 [22–24]. Stripecke et al. [25] were the first to report the transduction of primary human AMLs with lentiviral vectors (LV). We have subsequently designed LV to efficiently modify primary AML cells [24]. The immunostimulatory properties of CD80 and IL-2 are well characterised and there is accumulating in vitro [26–30], animal [31, 32] and clinical data [33–36] to suggest that they could be effective mediators of AML immunotherapy.
AML cells transduced with our vectors showed potent stimulatory capacity in lymphocyte proliferation assays. In addition, stimulated effector cells underwent expansion and showed increased levels of Th1 cytokines in culture supernatants [24]. We now report a small study showing that cultures stimulated with IL-2/CD80AML cells show an increased frequency of IFN-γ secreting cells, and increased MHC-dependent cytolytic activity against unmodified autologous AML blasts. In addition, we present preliminary evidence to suggest that the stimulated effectors preferentially respond to AML blasts compared to remission bone marrow cells.
Materials and methods
Patients and samples
Primary AML blasts were obtained from the peripheral blood of adult patients with high-count AML, at diagnosis and prior to initiation of chemotherapy. Peripheral blood mononuclear cells (PBMCs) were obtained from AML patients following chemotherapy and also from healthy volunteers. 15 AML samples were incubated with 51Cr as described below. Of 15 AML samples examined, ten labelled adequately and could be included in the study. Remission bone marrow was obtained from AML patients during cytogenetic and morphological remission. All patient samples were obtained after informed consent as approved by KCL Ethics Committee, in compliance with the principles of the Declaration of Helsinki (1964). Mutational analyses were carried out by the Cytogenetics and MRD laboratories, Department of Haematological Medicine, King’s College Hospital. FAB sub-typing and karyotyping were carried out by the Immunophenotyping Laboratory and Cytogenetics Units, respectively (KCH, London), for details see Table 1. All primary samples from patients or healthy donors were stored and anonymised in compliance with the regulations of the UK Human Tissue Act, 2005.
Cell culture
Acute myeloid leukaemia blasts, remission bone marrow cells and PBMCs were purified by Histopaque (Sigma) density gradient centrifugation and cryopreserved in X-VIVO 15 (BioWhittaker) with 10% DMSO and 50% Human AB serum (Sigma). Cryopreserved bone marrow samples from patients in remission were obtained from the Stem Cell Laboratory (King’s College Hospital, London). T cells were isolated from healthy donor PBMCs with a T cell Negative Isolation Kit (Dynal, Norway). All primary cells were cultured in X-VIVO 15 medium. AML cultures were supplemented with rhSCF (20 ng/ml) and rhIL-3 (10 ng/ml) (R&D Systems, UK) prior to and during lentiviral infection.
LV construction, virus production and titration procedures were carried out as previously described [24].
Genetic modification of AML cells
Cryopreserved AML blasts were thawed and cultured for 3 days in the presence of recombinant human SCF (20 ng/ml) and IL-3 (10 ng/ml) (R&D Systems, UK), prior to lentiviral (LV) infection. Target cells were washed and plated at 1 × 106 ml−1 and supplemented with 10 μg/ml DEAE dextran (Amersham Pharmacia). Aliquots of LV were thawed and infections performed overnight, at an MOI of 2–10 under standard culture conditions. The following day cells were washed twice and cultured for 72 h prior to analysis of IL-2 (ELISA) and CD80 expression (FACS) as previously described [24]. All samples showed a minimum of 40% CD80 positive cells after LV.CD80 or LV.IL-2/CD80 infection, and a minimum secretion of 1 ng IL-2/106cells/24 h after LV.IL-2 or LV.IL-2/CD80 infection. All samples showed lower expression of CD80 and IL-2 after infection with LV.IL-2/CD80 compared to LV.IL-2 or LV.CD80 infection (for results see Table 2). For extensive discussion of our gene transfer results please see Chan et al. [24].
Co-culture of effector cells and leukaemic cells
PBMCs (2 × 106) from healthy donors or AML patients in remission were seeded into 12-well plates, in a volume of 1 ml. Modified or unmodified AML blasts were prepared at 5 × 105 ml−1 and irradiated at 30 Gy. PBMC cultures received 1 ml of either medium only (unstimulated control), 5 × 105 unmodified AML, LV.CD80AML, LV.IL-2/CD80AML, or LV.IL-2 AML. In allogeneic AML patient cultures, the PBMCs and AMLs were from different patients. In some cases PBMC cultures were re-stimulated by the addition of 5 × 105 irradiated AML blasts (modified or unmodified) on day 7. At the end of the stimulation period, effector cells were washed and tested in functional assays (Cytotoxicity and ELISPOT assays). Nine autologous co-cultures were examined in this way; however, effector cells from three patients did not survive the culture period in adequate numbers, and hence could not be included in these assays.
Flow cytometry
FACS analysis of cell surface molecules was carried out on AML samples (MHCI-PE, MHCII-FITC, CD86-FITC, CD34-PE, CD54-FITC, and CD80-FITC) and effector cells (CD3-PE, CD4-PE, CD8-FITC, and CD56-FITC), all antibodies were obtained from Becton–Dickinson, Oxford, UK. Cells were washed and stained with antibody for 30 min at room temperature, in the dark (matched isotype controls were included for each sample). Stained cells were washed twice with HBSS, and resuspended in 300–500 μl HBSS. Flow cytometric analyses were carried out using a FACScalibur cytometer (BD Biosciences).
Chromium release assays
Unmodified AML cells were cultured for 3 days (as above) and washed twice to remove residual growth factors. Cells were labelled with 51Chromium (MP Biomedicals) for 1 h at 37°C, washed twice and seeded in X-VIVO 15 media in 96 well v-bottomed plates at a density of 1 × 104 per well. Effector cells were washed, resuspended in fresh X-VIVO media and added at various effector to target ratios. Plates were incubated for 4 h at 37°C, 5% CO2 and then centrifuged at 1,200 rpm. 100 μl aliquots of the supernatants were removed for scintillation counting. The fraction of cells lysed (% lysis) was calculated in relation to total and background levels of lysis in each assay. In experiments using MHC blocking antibodies, anti-HLA-DR,DP,DQ clone TU39 (BD Biosciences) and anti-HLA-ABC clone W6/32 (Serotec, Oxford) were added at a concentration of 20 μg/ml for 30 min prior to the addition of effector cells.
IFN-γ ELISPOT assays
Effector cells were washed twice and seeded at 2 × 105 per well into ELISPOT plates coated with IFN-γ capture antibody as per the manufacturer’s instructions (BD Biosciences, UK). Media alone or target cells at a density of 2 × 104 per well were added to wells. Target cells used were either unmodified AML cells or remission bone marrow cells. Plates were incubated for 24 h at 37°C, 5% CO2. Cells were then removed and the wells washed. IFN-γ spots were visualised according to the manufacturers’ instructions (Becton Dickenson) and counted by computer-assisted video image analysis using an AID ELISPOT reader (GmbH, Germany).
Results
Healthy donor or AML patient allogeneic effector cells are capable of killing unmodified AML cells after stimulation with IL-2/CD80 expressing AML cells
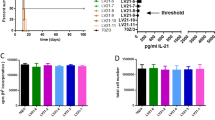
The capacity of the whole cell vaccine to stimulate lytic function was initially tested with healthy donor T cells. CD3+ T cells from a single healthy donor were stimulated in vitro with three different primary AML samples lentivirally transduced to express CD80 and IL-2, either singly or in combination. After a 2-week culture period, the stimulated cells were examined for their ability to kill unmodified AML cells from the same patient in a chromium release assay (Fig. 1a, b). Although the percentage lysis varied from sample to sample, a similar pattern was seen in each case. The CD3+ cells stimulated with unmodified AML cells showed low lytic activity. In comparison, T cells stimulated with the modified AML cells showed superior cytotoxicity at various effector to target ratios (Fig. 1b). T cells stimulated with IL-2/CD80AML cells elicited greater lysis (mean 23%) than T cells stimulated with AML cells expressing either CD80 (mean 14%) or IL-2 alone (mean 12%).

Lysis of primary AML blasts by allogeneic effector cells from a healthy donor, or from three AML patients in remission, after stimulation with modified AML cells. CD3+ cells from an unmatched healthy donor were co-cultured with media alone (unstimulated) or irradiated AML blasts (unmodified cells, LV.CD80, LVIL-2/CD80, LV.IL-2) for 2 weeks. Effector cells were then used in cytolytic assays with unmodified AML cells as targets. a The results with T cells from one healthy donor and three presentation AML samples at an effector to target ratio of 100:1. b A titration experiment performed with healthy donor T cells and AML cells from Patient 3 at different effector to target ratios. c PBMCs from three AML patients were stimulated with media alone or irradiated AML cells (unmodified, LV.CD80, LVIL-2/CD80, LV.IL-2) from an unrelated AML patient (Patient 2). Stimulated effector cells were then used in chromium release assays against unmodified AML cells from Patient 2 at an effector to target ratio of 50:1
We next investigated whether effector cells from AML patients were also capable of killing unmodified allogeneic AML cells (Fig. 1c). FACS analysis was carried out on AML patient PBMC samples prior to in vitro stimulation. All the patient samples contained CD4+, CD8+, and CD56+ cell subsets (Table 3). Accordingly, AML cells from Patient 2 were used to stimulate PBMCs from three unrelated AML patients in remission. After stimulation, the PBMCs were tested for their ability to kill unmodified AMLs from Patient 2 (Fig. 1c). Effector cells from two of the three patient cultures were able to cause lysis of the unmodified allogeneic AML cells after stimulation with modified AMLs. As with the healthy donor T cells, IL-2/CD80 AML cells stimulated a higher lytic response (mean 12%) compared to CD80 (mean 7%) or IL-2 (mean 8%) alone. Therefore, subsequent studies with autologous cultures were restricted to stimulations with IL-2/CD80 modified AML cells.
Stimulation of patient PBMCs with autologous IL-2/CD80 AML cells causes MHC-dependent cytolytic activity against unmodified AML cells
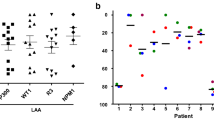
IL-2/CD80AML cells were used to prime patient PBMC which were subsequently examined for CD3, CD4, CD8, and CD56 staining and tested for lytic activity against unmodified autologous AML cells. Figure 2b shows a representative example of FACS staining of these cultures (Patient 3). Extensive phenotyping was not carried out, but the analysis suggested that CD4, CD8, and NK cells were present at the end of the culture period. Results of lytic assays obtained with PBMC from five patients in remission are shown in Fig. 2a. PBMCs from four of the five patients tested only caused lysis of unmodified AML cells (ranging from 5 to 24%) after prior priming with IL-2/CD80AML cells. In one case, PBMCs showed lytic activity after stimulation with media alone (10%) or unmodified cells (9%), but this was notably enhanced by IL-2/CD80AML cells (18%).

Cytotoxicity of patient PBMCs primed with autologous LV.IL-2/CD80 modified AML. PBMCs were obtained from AML patients in remission and stimulated with irradiated autologous AML cells (unmodified, LV.IL-2/CD80) or media only. Stimulated effector cells were analysed for surface markers and tested in cytotoxicity assays with unmodified autologous AML cells as targets. a Chromium release assays from five different AML patients. b The phenotype of the effector cells from Patient 3 on day 7 as determined by CD4, CD8, CD56, and CD3 surface staining. c Three chromium release assays repeated in the presence of MHC I and MHC II blocking antibodies
We repeated three of the cytolytic assays to address whether the killing was by MHC-dependent or MHC-independent mechanisms. Antibody-mediated blockade of MHC molecules on AML target cells partially inhibited the cytotoxicity of the stimulated PBMC in all three cases (Fig. 2c). The maximum blocking of lysis was seen with effector cells from Patient 3, with lysis by LV.IL-2/CD80AML-stimulated PBMCs being reduced by 64% by the presence of both blocking antibodies, indicating lysis is at least partly MHC dependent.
Stimulation of patient PBMC with autologous IL-2/CD80 AML cells increases the number of IFN-γ secreting effector cells
Effector cells from three AML patient cultures were also examined for IFN-γ release in response to unmodified autologous AML cells in ELISPOT assays. Figure 3 shows photographs of a representative well from each condition together with bar charts of triplicate values. A high frequency of IFN-γ spots was only detectable in response to unmodified AML cells after prior stimulation with IL-2/CD80 AML. In contrast, few spots were detectable with effectors pre-stimulated with media alone or unmodified AML cells, correlating with the chromium release assays shown in Fig. 2. The IL-2/CD80 AML-stimulated effector cells also showed a strong granzyme B response in ELISPOT assays; however, these wells were unquantifiable due to the high density of dots (data not shown).

IFN-γ secretion by patient PBMCs in response to autologous, unmodified AML cells. PBMCs obtained from three AML patients in complete remission were co-cultured with media alone (unstimulated), irradiated autologous AML cells (unmodified) or LV.IL-2/CD80AML for 7 days. Stimulated cells were then washed and incubated for 24 h in IFN-γ coated ELISPOT plates in the presence of unmodified autologous AML cells. A representative well is shown in each case, bar charts show mean values from triplicate wells
PBMCs from AML patients in remission primed by IL-2/CD80 AML cells show preferential recognition of unmodified leukaemic cells compared to normal haematopoietic cells
To investigate the specificity of the immune response, PBMCs isolated from two AML patients in remission were stimulated with IL-2/CD80 AML cells and subsequently tested against two different autologous secondary targets: AML blasts obtained at presentation and remission bone marrow cells (Fig. 4). In both patients tested, effector cells primed with IL-2/CD80 AML cells showed a higher frequency of IFN-γ secreting cells when the secondary target was unmodified AMLs compared to remission bone marrow.

IFN-γ response of effector cells from patients in remission to autologous leukaemic or normal haemopoietic cells after priming with LV.IL-2/CD80 AML cells. PBMCs from two AML patients in remission, either post-HSCT (Patient 3, a) or in chemotherapy-induced remission (Patient 7, b) were co-cultured with media only (unstimulated), unmodified AML or IL-2/CD80AML cells. After 1 week of co-culture 105 effector cells were incubated for 24 h in IFN-γ coated ELISPOT plates in the presence of no target, unmodified autologous AML cells or remission bone marrow (BM) cells. AML blasts were obtained from the original presentation sample, remission bone marrow cells were obtained during cytogenetic and morphological remission. Plots show the mean of triplicate wells
Discussion
Investigation of immune-mediated lysis of primary AML cells in vitro is hampered by the fact that the chromium release assay is not optimal for primary AML cells, as they often label poorly with the isotope. Therefore, it may not be possible to detect maximum levels of lysis. We investigated an alternative FACS-based killing assay [37], but in our hands, primary AML cells labelled poorly with CFSE. Therefore, despite its limitations, we used chromium release to investigate the killing of unmodified primary AML cells where possible. However, as many samples did not label adequately, this factor did reduce the number of samples available for these assays. Furthermore, effector cells from some AML patients did not survive in vitro culture. Therefore, only a small study using samples from a heterogeneous population of AML samples was possible. These variable in vitro characteristics of the samples could reflect a biological difference between patients.
We first examined the ability of primed effector cells to kill unmodified AML cells in an allogeneic setting. Using abundantly available, healthy donor T cells we were able to establish that AML cells modified with LV.IL-2.CD80 were more effective at promoting a cytolytic response than AML cells expressing either CD80 or IL-2 alone (Fig. 1a). This is encouraging, since it is well established that AML cells can impede cell-mediated immunity by a variety of mechanisms [19, 38, 39]. Since purified CD3+ cells were used for these experiments, it can be concluded that modified AML cells were directly stimulating effector cells in the absence of any other APCs. However, results with T cells from healthy donors are not directly comparable to those from AML patients, who often exhibit disease-related and treatment-induced immunosuppression [16]. Both NK and T cells from AML patients have been reported to show reduced in vitro effector function [13, 14]. In addition, the reconstitution of different immune cell subsets in AML patients who have received myeloablative chemotherapy progress at different rates, with NK cells appearing first, followed by CD8+ and lastly CD4+ cells [40]. We therefore tested the ability of AML patient PBMCs to lyse unmodified allogeneic AML cells after stimulation with modified AML cells. Of the three patient PBMCs tested, two were able to lyse an unrelated AML sample after priming with LV.IL-2/CD80-modified AML cells (Fig. 1c). This stimulation of effective killing by AML patient effector cells, albeit in an allogeneic setting, was highly encouraging. As before, the combination of CD80 and IL-2 was more effective than either transgene alone. We therefore limited the subsequent autologous assays to LV.IL-2/CD80-modified AML cells compared to unmodified AML cells.
All five patient samples tested showed increased lysis of autologous AML cells after priming with LV.IL-2/B7 modified cells compared to unmodified AMLs or media alone. Overall levels of lysis were not high (5–24%) but are encouraging, considering the immune suppression of most AML patients, the immunosuppressive qualities of AML cells and AML cell resistance to cell-mediated killing [13, 39, 41]. Our findings are in agreement with reports of T cell suppression in AML patients due to chemotherapy-induced cytopenia being reversible in vitro by anti-CD28 and IL-2 mediated stimulation [15].
Due to the availability of limited numbers of PBMCs from patients in remission, we were unable to purify CD3+ cells in sufficiently large numbers, and hence used unfractionated PBMC in autologous killing assays. Therefore, the cross-presentation of AML-derived antigens by professional APCs is a possibility. However, the use of PBMCs, rather than purified T cell subsets, is informative since it reflects the clinical situation, especially as AML patients can have atypical frequencies of lymphocyte subsets during immune reconstitution after chemotherapy and HSCT.
FACS analysis of the effector cell populations revealed CD56+CD3− populations which could be NK cells capable of MHC-independent cytotoxicity. However, inclusion of either MHC I and II blocking antibodies resulted in reduced cytotoxicity (Fig. 2b). All the AML samples used in this study were MHC I+ MHC II+ by FACS analysis (Table 2) and so could be susceptible to killing by both cytotoxic CD8+ and CD4+ T cells.
A potential danger of using a whole cell vaccine containing antigens present on both leukaemic and non-leukaemic cells is the generation of systemic allogeneic responses in transplanted patients, or autoimmunity against normal tissues in non-transplanted patients. Mutis et al. stimulated donor T cells with CD80-transfected AML cells and analysed the specificity of the resulting T cell population. They reported that the majority of T cell clones showed specificity for mHags HA-1 and HA-2 rather than LAAs [26].
We wished to investigate the specificity of the IL-2/CD80AML-generated response. Remission bone marrow samples were available for only two patients (one transplanted and one non-transplanted) and were used to compare the response of the IL-2/CD80AML-stimulated lymphocytes to a leukaemic and non-leukaemic target cell. Stimulated PBMCs from both AML patients showed a greater response to the presentation leukaemic cells than to the remission bone marrow cells. These higher responses to the leukaemic target, compared to normal haematopoietic cells, suggests the generation of an AML-specific response. Remission bone marrow is an adequate target for these assays as it is reflective of a potential site of GVHD and hence is clinically relevant. However, bone marrow contains a mixture of cells with different levels of differentiation. Therefore, our future studies will also compare the response to normal and leukaemic CD34+ cells, which share an immature phenotype.
Whilst encouraging, the data from this small study should be interpreted cautiously, since it may not be representative for all AML patients. Further experiments will be required to establish that this effect is reproducible in a large cohort of AML patients.
References
Goldstone AH, Burnett AK, Wheatley K, Smith AG, Hutchinson RM, Clark RE (2001) Attempts to improve treatment outcomes in acute myeloid leukaemia (AML) in older patients: the results of the United Kingdom Medical Research Council AML11 trial. Blood 98:1302
Hann IM, Stevens RF, Goldstone AH, Rees JK, Wheatley K, Gray RG, Burnett AK (1997) Randomized comparison of DAT versus ADE as induction chemotherapy in children and younger adults with acute myeloid leukaemia. Results of the Medical Research Council’s 10th AML trial (MRC AML10). Adult and Childhood Leukaemia Working Parties of the Medical Research Council. Blood 89:2311
Chan L, Hardwick NR, Guinn BA, Darling D, Gaken J, Galea-Lauri J, Ho AY, Mufti GJ, Farzaneh F (2006) An immune edited tumour versus a tumour edited immune system: prospects for immune therapy of acute myeloid leukaemia. Cancer Immunol Immunother 55:1017
Molldrem JJ (2006) Vaccination for leukaemia. Biol Blood Marrow Transplant 12:13
Kolb HJ, Schmid C, Barrett AJ, Schendel DJ (2004) Graft-versus-leukemia reactions in allogeneic chimeras. Blood 103:767
Andersen MH, Svane IM, Kvistborg P, Nielsen OJ, Balslev E, Reker S, Becker JC, Straten PT (2005) Immunogenicity of Bcl-2 in patients with cancer. Blood 105:728
Greiner J, Ringhoffer M, Taniguchi M, Schmitt A, Kirchner D, Krahn G, Heilmann V, Gschwend J, Bergmann L, Dohner H, Schmitt M (2002) Receptor for hyaluronan acid-mediated motility (RHAMM) is a new immunogenic leukemia-associated antigen in acute and chronic myeloid leukemia. Exp Hematol 30:1029
Greiner J, Ringhoffer M, Taniguchi M, Hauser T, Schmitt A, Dohner H, Schmitt M (2003) Characterization of several leukaemia-associated antigens inducing humoral immune responses in acute and chronic myeloid leukaemia. Int J Cancer 106:224
Guinn BA, Gilkes AF, Woodward E, Westwood NB, Mufti GJ, Linch D, Burnett AK, Mills KI (2005) Microarray analysis of tumour antigen expression in presentation acute myeloid leukaemia. Biochem Biophys Res Commun 333:703
Scheibenbogen C, Letsch A, Thiel E, Schmittel A, Mailaender V, Baerwolf S, Nagorsen D, Keilholz U (2002) CD8 T-cell responses to Wilms tumor gene product WT1 and proteinase 3 in patients with acute myeloid leukaemia. Blood 100:2132
Lim SH, Worman CP, Jewell AP, Goldstone AH (1991) Cellular cytotoxic function and potential in acute myelogenous leukaemia. Leuk Res 15:641
Morikawa K, Nakano A, Oseko F, Morikawa S (1989) Depressed natural killer (NK) function in blood and marrow is related to the decrease in CD11+ cells in acute leukaemia. Jpn J Med 28:485
Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, Gastaut JA, Pende D, Olive D, Moretta A (2002) Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukaemia. Blood 99:3661
Buggins AG, Hirst WJ, Pagliuca A, Mufti GJ (1998) Variable expression of CD3-zeta and associated protein tyrosine kinases in lymphocytes from patients with myeloid malignancies. Br J Haematol 100:784
Wendelbo O, Nesthus I, Sjo M, Paulsen K, Ernst P, Bruserud O (2004) Functional characterization of T lymphocytes derived from patients with acute myelogenous leukaemia and chemotherapy-induced leukopenia. Cancer Immunol Immunother 53:740
Mackall CL (2000) T-cell immunodeficiency following cytotoxic antineoplastic therapy: a review. Stem Cells 18:10
Nguyen S, Dhedin N, Vernant JP, Kuentz M, Al Jijakli A, Rouas-Freiss N, Carosella ED, Boudifa A, Debre P, Vieillard V (2005) NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 105:4135
Whiteway A, Corbett T, Anderson R, Macdonald I, Prentice HG (2003) Expression of co-stimulatory molecules on acute myeloid leukaemia blasts may effect duration of first remission. Br J Haematol 120:442
Chamuleau ME, Souwer Y, Van Ham SM, Zevenbergen A, Westers TM, Berkhof J, Meijer CJ, van de Loosdrecht AA, Ossenkoppele GJ (2004) Class II-associated invariant chain peptide expression on myeloid leukemic blasts predicts poor clinical outcome. Cancer Res 64:5546
Buggins AG, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, Hirst WJ (2001) Microenvironment produced by acute myeloid leukaemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J Immunol 167:6021
Orleans-Lindsay JK, Barber LD, Prentice HG, Lowdell MW (2001) Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function—implications for the adoptive immunotherapy of leukaemia. Clin Exp Immunol 126:403
Hirst WJ, Buggins A, Darling D, Gaken J, Farzaneh F, Mufti GJ (1997) Enhanced immune costimulatory activity of primary acute myeloid leukaemia blasts after retrovirus-mediated gene transfer of B7.1. Gene Ther 4:691
Buggins AG, Lea N, Gaken J, Darling D, Farzaneh F, Mufti GJ, Hirst WJ (1999) Effect of costimulation and the microenvironment on antigen presentation by leukemic cells. Blood 94:3479
Chan L, Hardwick N, Darling D, Galea-Lauri J, Gaken J, Devereux S, Kemeny M, Mufti G, Farzaneh F (2005) IL-2/B7.1 (CD80) fusagene transduction of AML blasts by a self-inactivating lentiviral vector stimulates T cell responses in vitro: a strategy to generate whole cell vaccines for AML. Mol Ther 11:120
Stripecke R, Cardoso AA, Pepper KA, Skelton DC, Yu XJ, Mascarenhas L, Weinberg KI, Nadler LM, Kohn DB (2000) Lentiviral vectors for efficient delivery of CD80 and granulocyte–macrophage- colony-stimulating factor in human acute lymphoblastic leukaemia and acute myeloid leukaemia cells to induce antileukemic immune responses. Blood 96:1317
Mutis T, Schrama E, Melief CJ, Goulmy E (1998) CD80-Transfected acute myeloid leukaemia cells induce primary allogeneic T-cell responses directed at patient specific minor histocompatibility antigens and leukaemia-associated antigens. Blood 92:1677
Koya RC, Kasahara N, Pullarkat V, Levine AM, Stripecke R (2002) Transduction of acute myeloid leukaemia cells with third generation self-inactivating lentiviral vectors expressing CD80 and GM-CSF: effects on proliferation, differentiation, and stimulation of allogeneic and autologous anti-leukaemia immune responses. Leukaemia 16:1645
Notter M, Willinger T, Erben U, Thiel E (2001) Targeting of a B7-1 (CD80) immunoglobulin G fusion protein to acute myeloid leukaemia blasts increases their costimulatory activity for autologous remission T cells. Blood 97:3138
Orleans-Lindsay JK, Deru A, Craig JI, Prentice HG, Lowdell MW (2003) In vitro co-stimulation with anti-CD28 synergizes with IL-12 in the generation of T cell immune responses to leukaemic cells; a strategy for ex-vivo generation of CTL for immunotherapy. Clin Exp Immunol 133:467
Bruserud O, Ulvestad E (2000) Cytokine responsiveness of mitogen-activated T cells derived from acute leukaemia patients with chemotherapy-induced leukopenia. J Interferon Cytokine Res 20:947
Dunussi-Joannopoulos K, Weinstein HJ, Nickerson PW, Strom TB, Burakoff SJ, Croop JM, Arceci RJ (1996) Irradiated B7-1 transduced primary acute myelogenous leukaemia (AML) cells can be used as therapeutic vaccines in murine AML. Blood 87:2938
Boyer MW, Vallera DA, Taylor PA, Gray GS, Katsanis E, Gorden K, Orchard PJ, Blazar BR (1997) The role of B7 costimulation by murine acute myeloid leukaemia in the generation and function of a CD8+ T-cell line with potent in vivo graft-versus-leukaemia properties. Blood 89:3477
Meloni G, Foa R, Vignetti M, Guarini A, Fenu S, Tosti S, Tos AG, Mandelli F (1994) Interleukin-2 may induce prolonged remissions in advanced acute myelogenous leukaemia. Blood 84:2158
Meloni G, Trisolini SM, Capria S, Torelli GF, Baldacci E, Torromeo C, Valesini G, Mandelli F (2002) How long can we give interleukin-2? Clinical and immunological evaluation of AML patients after 10 or more years of IL2 administration. Leukaemia 16:2016
Hicks C, Cheung C, Lindeman R (2003) Restimulation of tumour-specific immunity in a patient with AML following injection with B7-1 positive autologous blasts. Leuk Res 27:1051
Zhang WG, Liu SH, Cao XM, Cheng YX, Ma XR, Yang Y, Wang YL (2005) A phase-I clinical trial of active immunotherapy for acute leukaemia using inactivated autologous leukaemia cells mixed with IL-2, GM-CSF, and IL-6. Leuk Res 29:3
Jedema I, van der Werff NM, Barge RM, Willemze R, Falkenburg JH (2004) New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood 103:2677
Dermime S, Mavroudis D, Jiang YZ, Hensel N, Molldrem J, Barrett AJ (1997) Immune escape from a graft-versus-leukaemia effect may play a role in the relapse of myeloid leukemias following allogeneic bone marrow transplantation. Bone Marrow Transplant 19:989
Lehmann C, Zeis M, Schmitz N, Uharek L (2000) Impaired binding of perforin on the surface of tumor cells is a cause of target cell resistance against cytotoxic effector cells. Blood 96:594
Ohnishi K, Yamanishi H, Naito K, Utsumi M, Yokomaku S, Hirabayashi N, Ohno R (1998) Reconstitution of peripheral blood lymphocyte subsets in the long-term disease-free survivors of patients with acute myeloblastic leukaemia. Leukaemia 12:52
Buzyn A, Petit F, Ostankovitch M, Figueiredo S, Varet B, Guillet JG, Ameisen JC, Estaquier J (1999) Membrane-bound Fas (Apo-1/CD95) ligand on leukemic cells: a mechanism of tumor immune escape in leukaemia patients. Blood 94:3135
Acknowledgments
We are grateful to Dr Linda Barber for critical feedback on this manuscript. This work was funded by The Leukaemia Research Fund and The Elimination of Leukaemia Fund. The authors also acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hardwick, N., Chan, L., Ingram, W. et al. Lytic activity against primary AML cells is stimulated in vitro by an autologous whole cell vaccine expressing IL-2 and CD80. Cancer Immunol Immunother 59, 379–388 (2010). https://doi.org/10.1007/s00262-009-0756-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-009-0756-x