
Abstract
The concept of using replicating oncolytic viruses in cancer therapy dates to the beginning of the twentieth century. However, in the last few years, an increasing number of pre-clinical and clinical trials have been carried out with promising preliminarily results. Novel, indeed, is the suggestion that viral oncolytic therapy might not operate exclusively through an oncolysis-mediated process but additionally requires the “assistance” of the host’s immune system. Originally, the host’s immune response was believed to play a predominant obstructive role against viral replication, hence limiting the anti-tumor efficacy of viral vectors. Recent data, however, suggest that the immune response may also play a key role in promoting tumor destruction in association with the oncolytic process. In fact, immune effector pathways activated during oncolytic virus-induced tumor rejection seem to follow a similar pattern to those observed when the broader phenomenon of immune-mediated tissue-specific rejection occurs in other immune-related pathologies. We recently formulated the “Immunologic Constant of Rejection” hypothesis, emphasizing commonalties in transcriptional patterns observed when tissue-destruction occurs: whether with a favorable outcome, such as in tumor rejection and pathogen clearance; or a destructive one, such as in allograft rejection or autoimmunity. Here, we propose that a similar mechanism induces clearance of virally infected tumors and that such a mechanism is primarily dependent on innate immune functions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Historical synopsis of viral oncolytic treatment
Oncolytic viruses were first noticed in the early twentieth century when some cancer patients were noted to undergo tumor regression after systemic viral infections [1]. A leukemia patient, for instance, was reported to undergo remission after acute infection with influenza virus [2].
The realization that some viruses with lytic properties selectively propagate in and colonize tumors while leaving healthy tissues unharmed [3] originated the concept of “oncolytic viruses”. These viruses were considerate attractive candidates for the treatment of established tumors. Moreover, while a wide array of replication-deficient oncolytic viruses has been used in the past, recent studies have clearly shown that these constructs lack efficacy when compared to other replication-competent models [4].
The mutant adenovirus ONYX-015, which carries a deletion for E1B-55K, was the first replication-competent modified virus that displayed anti-cancer effects in humans [5], and was originally believed to target p53-deficient cancer cells. The use of this genetically engineered virus as a therapeutic agent has progressed to phase III clinical trials only 4 years after its first application in patients [6].
It is important to consider that the safe administration of oncolytic viruses in humans depends on an exclusive tropism for cancer cells. This has been achieved, among other methods, by the disruption of non-essential viral genes in viruses such as in HSV [7], adenovirus [8] and vaccinia [9] that, for unclear reasons, alters the replicative capacity of viruses in a tissue-specific fashion.
Oncolytic therapy with vaccinia virus (VACV)
VACV is a promising candidate poxvirus for oncolytic therapy due to its extensive use in humans for vaccination worldwide against smallpox. This experience has clearly been demonstrated it to be safe. In addition, VACV displays several benefits when compared to other oncolytic viruses. One of the biggest advantages is that VACV’s genome of about 200 kb is relatively permissive to the insertion of foreign genes up to 25 kb length [10], which have been used to modulate the in situ function of the virus, the infected cells and bystander host cells that are reacting to the intra-tumoral infectious process. This has been achieved through the expression of immune-modulatory genes, marker genes, therapeutic proteins or drug-converting enzymes. Another advantageous feature of VACV is the susceptibility of almost all cells within a tumor to infection with this virus, and VACV infection not only induces cell lysis but it also induces an associated, and in some cases preceded, activation of local cytotoxic T cells (CTLs) and natural killer cells (NK) responses. At the same time, with the exception of immune compromised patients or those with certain skin conditions such as eczema, VACV does not cause serious pathologies in humans and it has been shown to naturally and selectively propagate in tumor cells.
To enhance the natural tumor tropism of VACV, Kirn et al. [11] deleted the B18R gene, which encodes a protein that neutralizes type I Interferons (IFNs), producing a multi-functional and highly tumor-specific oncolytic vaccinia virus. Others [12] have designed a highly attenuated thymidine kinase- and vaccinia growth factor-depleted virus strain (vvDD-GFP) with enhanced anti-tumor efficacy. We recently introduced a novel VACV strain (GLV-1h68) derived from the LIVP progenitor strain that was modified by insertion of three expression cassettes (Renilla luciferase-Aequorea green fluorescent fusion protein, β-galactosidase and β-glucoronidase) into the F14.5L, J2R and A56R loci of the viral genome, respectively. Because of its light emitting properties, GLV-1h68 can be used simultaneously as an imaging tool to detect malignant cells in the body while exerting its oncolytic effects [13].
Innate immune reactions to pathogenic vaccinia virus infection
Host cells possess the ability to sense viral infections through specific membrane bound or soluble intracellular pattern recognition receptors. Their ligands, pathogen-associated molecular patterns, trigger signaling cascades which ultimately lead to the production of type I IFNs and other cytokines. These innate immunity mechanisms protect the cells from uncontrolled virus spread while the adaptive immune response fully matures.
Recent studies have also demonstrated that the immune system may utilize ancestral autophagy mechanisms that have generally been adopted as a primary defense to battle microorganisms invading infected cells. The underlying mechanisms are still to be elucidated, however, it is clearly accepted that the anti-apoptotic function of autophagy provides protection against virus-induced pathologies [14].
Immune reactions against VACV infections have been studied and appear to differ according to the route of VACV administration. Intra-nasal infections of BALB/c mice with VACV strain Western Reserve induced early viral replication in the upper respiratory tract and lung that was associated with the infiltration of inflammatory cells into the lungs up to 15 days following infection. This infiltration included predominantly macrophages and T lymphocytes as well as the expression of several CCL chemokines (3, 2 and 11) and CXCL chemokines (1 and 2/3) [15]. Intra-dermal infection of the ears of BALB/c and C57BL/6 mice is followed by recruitment of macrophages, granulocytes and predominantly T cell receptor (TCR)-γδ-expressing T lymphocytes. This primary response is secondarily followed by a large infiltration of CD4+ and CD8+ T cells [15, 16]. This was consistent with findings observed with the intra-dermal and intra-nasal route of administration. Selin et al. [17] proposed a role for IFN-γ-producing γδ-T cells following VACV delivery in the peritoneal cavity of C57BL/6 mice. Interestingly, uninfected β-TCR knock out mice harbored a significant number of VACV-specific γδ-T cells, which rapidly expanded in response to VACV infection and secreted IFN-γ while deploying increased cytotoxic activity. Thus, IFN-γ production is critical for the clearance of acute VACV infection [18, 19]. VACV, however, has evolved to protect itself from the host’s anti-viral response. Like many other viruses, VACV encodes genes that can interfere with the host’s innate immune defense [20]. VACV induces many intra- and extra-cellular proteins that can inhibit IFN-γ anti-viral effects. A good example is the product of the viral gene B8R, a soluble-IFN-γ receptor like-molecule, which binds specifically human and not mouse IFN-γ [21, 22], and results in a species-specific immune evasion mechanisms in its natural host.
Whereas, a type II IFN-mediated immune response is responsible for the activation of the acute inflammatory response associated with oncolytic therapy and is central to the activation of cell-mediated immunity [23], both type I and type II IFNs are equally important in the containment of poxvirus infections [19, 24, 25]. In addition to its direct antiviral effects, IFN-α orchestrates a wide array of immune regulatory and other effects [26] including the regulation of NK cell activation [27, 28]. The direct signaling of NK cells, but not dendritic cells (DCs) through IL-15 secretion [29] seems to be required for the activation and exhibition of the effector functions which lead to VACV clearance both in vitro and in vivo [30].
Cancer immunotherapy with vaccinia virus as an immunization agent
It is generally accepted that more than one genetic alteration is needed to transform normal cells into cancerous ones and to begin independent growth. It is also well accepted that the early evolution of cancer leads to the changing and adapting of phenotypes that allows tumor escape from immune surveillance through a mechanism referred to as immune editing [31–33]. Thus, tumors adapt to immune responses by down-modulating their antigenic properties and by producing factors that dampen the immune response. This creates a microenvironment in which inflammation predominantly fosters tumor growth [34–36]. Moreover, although cancer cells express tumor-associated-antigens (TAAs) that can be naturally recognized by the host’s immune system [37, 38], their immunogenic potential is, in normal conditions, insufficient to induce effective cytotoxic cell activation due to lack of co-stimulatory signals generally provided in other immune-mediated phenomena such as by the expression of pathogen associated patterns [39]. In that context, the total loss or decrease in expression of HLA class I molecules has been reported for various cancer cells and this down-regulation eventually prevents tumor regression since this renders cancer cells insensitive to CTLs. In melanoma patients, low HLA class I antigen expression has been linked to poor clinical outcome and lesions that progress after immunotherapy [40, 41].
Thus, contrary to cells infected by lytic viruses, cancer cells survive in an immune quiescent microenvironment in which chronic inflammatory processes are not sufficient to induce their elimination. This is an important consideration when evaluating the immune effects that an oncolytic virus infection can produce and how it may alter the microenvironment.
Several clinical approaches have been tested to make a tumor “visible” to the immune system and thus to direct an immune response against it. Although cancer chemotherapy has usually been considered to be immunosuppressive, recent data suggest that some chemotherapeutic agents might trigger an anticancer immune response similar to that which can be achieved with oncolytic therapy [42, 43]. Viral vectors have been used to deliver and express tumor antigens with the aim of making the tumor cells more immunogenic [44, 45]. Virally infected cells provide the danger signals which are sensed by cells of the innate immune system. Once triggered by the presence of the pathogen, DCs, NK cells, macrophages, neutrophils, basophils, eosinophils and mast cells secrete cytokines and chemokines, which either lead to the direct killing of the pathogen-infected cell or lead to the recruitment of other inflammatory cells belonging to the adaptive immune response [46]. The concomitant activation of the immune system is one of the most important goals in the use of viral vectors as cancer vaccines. Thus, although these viruses have no oncolytic activity and have no specific cancer cell tropism, they can induce anti-cancer effects through the activation of the immune responses against TAAs that would be otherwise indolent due the lack of immune cell co-stimulation in the absence of a viral infection.
While the decision to select the optimal TAA is influenced by several factors beyond the scope of this review, the selection of the viral delivery system addresses more basic issues. Tumor-specific tropism, replication and induction of long-lasting, and potent cellular and humoral responses paired with safety for the patient are all considerations which places VACV among the most promising potential vectors [47]. A study conducted in the United Kingdom described the use of Modified Vaccinia Ankara (MVA) virus to deliver the tumor antigen 5T4 in colorectal cancer patients. 5T4 oncofetal antigen is a non-mutated self-antigen similar to carcinoembryonic antigen according to the classification system described by Amato et al. [47]. In a preliminary phase II study, it was observed that 6 out of 11 patients experienced a complete or partial response to the MVA-5T4 (TroVax®) administration [48]. More recently, the same group reported their experience in more than 200 patients who received over 700 doses of vaccine and immune responses were observed in approximately 95% of patients [47].
Kaufman et al. [49] reported the administration of two genetically engineered poxvirus family members: VACV and Fowlpox virus. Both constructs carried the TAAs carcinoembryonic antigen and mucin-1 and were administered to pancreatic cancer patients. Vaccination was well tolerated and five out of eight patients displayed antigen specific T-cell responses after VACV injection.
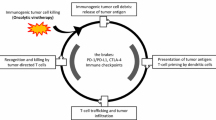
To stimulate strong T-cell responses against weak TAAs, co-stimulatory signals are needed [50, 51]. Stanford et al. [46] suggested that the achievement of immune-mediated tumor regression requires three criteria to be fulfilled: (a) the generation of a large frequency of high avidity tumor reactive-T-cells, (b) the trafficking of tumor-reactive T-cells to the tumor site, and (c) sustained T-cell activation within the tumor. Similarly, we have long argued that the localization and activation of T-cells at the tumor site is the key factor missing for successful immune-mediated tumor destruction [51–53] and that immunotherapy works primarily through the activation of potent innate immune responses within the tumor microenvironment, which could cause a switch from a lingering and chronic inflammatory process to an acute one [54–57]. Thus, poor clinical response rates in clinical trials with cancer vaccines [58] may be due to the failure to achieve the third criteria and secondary to tumor-evolved mechanisms that dampen adaptive responses and inhibit full activation of T-cells, the requirements of which have not been sufficiently characterized [53].
The expression of immunosuppressive factors like IL-10 or TGF-β by tumor-associated immune cells, as well as the T-regulatory cell mediated suppression of CTL proliferation and effector function may prevent a successful outcome [46]. It is in this realm that oncolytic therapy may play an important role in facilitating the activation of vaccine-induced immune responses by providing powerful co-stimulation at the tumor site [53].
Recent data suggest that angiogenesis inhibitors may have a beneficial effect for clinical outcome. Tumor cells tend to produce angiogenic growth factors and factors that suppress the expression of endothelial cell adhesion molecules that are necessary for interaction with leukocytes. Agents such as anti-VEGF antibodies, endostatin and anginex share the ability to normalize the expression of adhesion molecules and thus stimulate leukocyte infiltration and make the tumor more vulnerable to the immune system [59].
Innate immune responses have been shown to induce tumor rejection without necessarily requiring the presence of adaptive immune responses [60]. Indeed, it has been argued that adaptive cytotoxic T cell responses may act primarily as “helpers” to promote powerful activation of innate immune effectors such as NK cells [61]. In humans, local injection of Toll-like receptor agonists or adenoviral vectors can induce the regression of basal cell carcinomas, cutaneous lymphomas, actinic keratosis and leukemias without evidence of B or T-cell participation [56, 62–64] although pre-clinical models suggest a potential role of Toll-like receptor signaling activation in expanding adaptive immune responses [65]. We have recently argued that adaptive immune responses provide specificity but are not necessarily sufficient to induce cancer rejection unless other components of the innate immune response are activated at the same time [57]. It is in this interface that VACV (and more broadly viral oncotropic viral vectors) may play a key role in tumor immunology as discussed in the next section.
Oncolytic therapy with VACV and tumor rejection in the context of an innate immune response
The innate immune response initially stimulated by the virally infected cells and/or the VACV itself is directed automatically against the infected tumor cells and we suggest that this is part of the mechanism leading to tissue destruction by oncolytic therapy. A critical parameter is the likelihood of viral localization and replication at the tumor site before the pathogen can be neutralized by the host’s immune system. Extra-cellular enveloped virus (EEV), a VACV form that naturally occurs early after infection, leads to rapid cell-to-cell spread before cell lysis occurs. EEV particles possess a host cell-derived lipid bi-layer which contains anti-complement proteins, hence providing protection against immune-mediated clearance. Infections in murine models with EEV-enhanced VACV strains result in improved anti-tumor effects due to their enhanced ability to spread and replicate in distant tumor parts and to be more resistant to neutralizing antibodies [66].
We have generated the efficiently replicating VACV GLV-1h68 strain, which leads to regression of GI-101A breast cancer xenografts in 95% of cases after intravenous delivery of 1 × 107 viral particles. Insertion of three foreign expression cassettes into the F14.5L, J2R, and A56R loci of the parental LIVP genome, expressing Renilla luciferase-Aequorea green fluorescent protein (RUC-GFP) fusion, β-galactosidase, and β-glucuronidase, respectively, resulted in a replicating, oncolytic VACV strain with increased tumor specificity and hence less systemic toxicity [13].
Transcriptional analysis of mouse xenografts using a mouse-specific platform to identify the host’s response genes revealed the activation of innate immune mechanisms in regressing GI-101A tumors compared to non-infected control tumors [13]. Up-regulation of pro-inflammatory chemokine ligands such as CCL2, CCL17-19, CCL12, CXCL9, CXCL10 and CXCL12 was seen together with an increase in interleukin (IL), and chemokine receptors (IL13R, IL18, and CCR2) transcripts. Additionally, a significant activation of Interferon-stimulated genes ISGs (Ifi202b, Ifi203, Ifi204, Ifi205, Ifi35, Ifi44, Ifi47, Ifih1, Ifit1, Ifit2, Ifit3, Ifitm3, Igtp, Iigp1, Isgf3g) was observed in association with increased STAT1 and Interferon-regulatory factor (IRF)-7. This strongly suggested that type I IFNs are critically involved in the process. Finally, immunohistochemistry of VACV-infected, regressing xenografts showed an intense peri- and intra-tumoral infiltration of mononuclear cells, which was confirmed by the up-regulation of CD69, CD48, CD52, and CD53 seen in the host’s gene expression arrays. These markers are expressed on activated T-cells, NK cells, macrophages, granulocytes and DCs, and are associated with leukocyte activation and NK cytolytic function [13]. We concluded that tumor rejection induced by oncolytic viruses in this xenograft model is at least in part mediated through activation of innate immune mechanisms which correlate with the level of viral replication and precede tumor regression.
In a recent study, we compared GLV-1h68-infected GI-101A xenografts which were sensitive to oncolytic therapy to GI-101A xenografts from non-infected animals and to HT-29 colon cancer xenografts that do not respond to oncolytic therapy [67]. Moreover, we evaluated gene expression profiles of the oncolytic interaction by adopting organism-specific microarray platforms to simultaneously monitor gene expression changes in the tumor microenvironment. We applied 36k whole genome human arrays to test for alterations in the human cancer cells; 36k whole genome mouse arrays to examine the host’s infiltrating stromal cells and lastly; custom-made 1K VACV arrays to characterize changes in viral transcription pattern.
Interestingly, human transcript analysis revealed no differences in non-responding, infected HT-29 tumors compared to control tumors. The expression of only a limited set of genes was altered after GLV-1h68 inoculation in regressing GI-101A xenografts. Most of the transcriptional changes that were observed in the infected responding tumors at a time when cell death had not yet occurred revealed a profound down-regulation of genes associated with cellular metabolic processes. These changes reflected the shut down of cancer cell metabolism due to VACV infection. Most of the few up-regulated transcripts in regressing GI-101A cells infected with VACV were associated with the activation of the innate immune mechanisms, but further sequence analysis showed that the majority represented the host’s mouse cell transcripts that cross-hybridized onto human arrays.
The most interesting insights of this study were gained after analysis of the mouse expression arrays that represented the host’s infiltrating cells. Infected, non-responsive HT-29 tumors did not show significant changes in gene expression compared to HT-29 tumors from non-infected control animals. On the contrary, a large number of genes were up-regulated in the GI-101A tumors after GLV-1h68 delivery compared to the non-infected GI-101A xenografts. Further analysis discovered a significant enrichment of immune-related genes. Among these immune-regulated genes, ISGs and other IFN signaling genes represented the most up-regulated canonical pathways both at an early time points when tumors were still continuing to grow in size (21 days post-infection) and later (42 days post-infection) when tumor rejection had started. This suggested that strong immune activation precedes tumor necrosis and, therefore, is unlikely secondary to the death of cancer cells, but rather is related to cancer cell infection. We also observed the up-regulation of IL-18 and IL-18 binding proteins, both of which played a dominant role early in infection, whereas IL-15 became the predominant cytokine expressed at later stages. Among CXCL chemokines, CXCL9, CXCL11 and CXCL12 were strongly expressed in regressing GI-101A xenografts together with CCL5 and CCL9 [67].
Based on these findings, we concluded that, in this immune deficient mouse model, the activation of innate immune responses might be sufficient to lead to tumor regression in cooperation with the viral oncolytic process. Further analysis of the genes activated upon tumor rejection in this mouse model leads to the integration of the results in the context of a much broader phenomenon that we recently described as the immunologic constant of rejection (ICR) [57].
The immunologic constant of rejection
In 1969, Salk proposed the question of whether chronic infections, allograft rejection, autoimmune disorders and cancers belong to a common phenomenon that he termed the “delayed allergy reaction” [68]. The underlying mechanisms of these pathologies are clearly variable and distinct from each other. Infectious diseases like hepatitis C virus (HCV) infections become chronic if the pathogen is not cleared acutely; the latter process occurs rarely in humans [69]. Allograft transplant rejection can be controlled only through immune suppression because the broad antigenic differences between the allograft and host can trigger a strong immune response. Whereas, both allograft rejection and pathogen clearance represent an immune reaction against non-self structures, the immune system can also attack “self” tissues if the discrimination between self and non-self fails, as observed in autoimmunity. Immune responses against tumors fit self or non-self discrimination, as tumor cells are derived from normal progenitor cells and mostly express non-mutated TAAs [70]. However, some cancers display mutated antigens [71] that are unique to tumor cells and can be recognized as non-self by the immune system.
Even though the underlying triggering mechanisms differ among distinct immune pathologies, we postulated that the final outcome defined as tissue-specific destruction follows a common effector pathway which we called the “immunologic constant of rejection”. We formulated four axioms that summarize the phenomenon: (1) tissue specific destruction does not necessarily occur because of non-self recognition but also occurs against self or quasi-self; (2) the requirements for the induction of a cognate immune response differ from those are necessary for the activation of an effector one; (3) although the prompts leading to tissue specific destruction vary in distinct pathologic states, the effector immune response converges into a single mechanism; and (4) adaptive immunity participates as a tissue-specific trigger, but it is not always sufficient or necessary [57].
The ICR theory was formulated based on studies conducted in humans but perfectly supports our findings in the xenograft model described above. Athymic mice harboring GI-101A xenografts mount an innate response and are able to reject the tumors after systemic delivery of the oncolytic VACV GLV-1h68 [13]. According to the ICR hypothesis, common effector pathways consisting of activation of ISGs and immune effector functions such as cell-mediated toxicity were all up-regulated in regressing GI-101A tumors in the mouse transcriptome suggesting a local innate immune response. No such immune reaction was seen in HT-29 tumors which corresponded with continued growth of the respective xenografts. Furthermore, responding tumors showed an activation of a distinct subset of macrophage-associated signatures and were highly infiltrated by MHC class II positive cells in the intra- and peri-tumoral compartment [67]. Interestingly, after being initially vaccinated with 5 × 106 pfu of GLV-1h68 responding GI-101A tumors showed much higher viral titers than persistent HT-29 tumors. It appears that in vivo viral titers are key players in determining the intensity of the immune response and, as a consequence, treatment outcome (Fig. 1).

a HT-29 xenografts did not respond to oncolytic therapy with GLV-1h68, an attenuated vaccinia virus strain. No immune reaction was seen in HT-29 tumors which corresponded with continued growth of the respective xenografts. b After being initially vaccinated with the same dose of GLV-1h68 responding GI-101A tumors showed much higher viral titers than persistent HT-29 tumors. Responding tumors were highly infiltrated by MHC class II positive cells in the intra- and peri-tumoral compartment and presented a distinct activation of pro-inflammatory genes which lead to immune-mediated tissue destruction
While the weight and timing of the oncolytic effects compared to the activation of the innate immunity needs to be elucidated, it seems clear that adaptive immunity is not required to reject tumors during oncolytic therapy. This is not totally surprising as similar findings were observed in experimental tumor regression models [60]. During oncolytic therapy, the presence of the virus selectively in the tumor cells probably induces an immune-stimulation within the target organ that is sufficient to bypass the need for the specificity provided by TAA-specific T-cells. The potent pro-inflammatory viral infection in these cases is sufficient to induce a switch from a chronic inflammatory status to an acute one and leads to tissue specific destruction.
Conclusion
In summary, it appears that oncolytic therapy with VACV is associated in experimental models with powerful activation of immune responses within the tumor microenvironment. The ultimate role of the immune response in determining tumor rejection compared with the direct oncolytic process needs to be further evaluated by immune-depletion experiments. However, the strong association between viral replication in cancer cells and activation of immune responses suggests that oncolytic viruses characterized by specific tropism for cancer cells may be exploited not only as therapeutic tools in the context of classical oncolytic therapy but also as adjuvant in the context of other immunotherapy strategies, including the active specific immunization where, we believe, the limit to successful tumor eradication is due to the lack of stimulation of vaccine-induced T-cells in the target tissue [53]. This hypothesis needs to be further evaluated in future clinical trials that follow a systematic approach [72], but it is likely that oncolytic therapies will play a critical role in identifying important mechanisms leading to immune-mediated tissue-specific rejection.
References
Sinkovics J, Horvath J (1993) New developments in the virus therapy of cancer: a historical review. Intervirology 36:193–214
Dock G (1904) The influence of complicating diseases upon leukaemia. Am J Med Sci 127:563
Parato KA, Senger D, Forsyth PA, Bell JC (2005) Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer 5:965–976
Vaha-Koskela MJ, Heikkila JE, Hinkkanen AE (2007) Oncolytic viruses in cancer therapy. Cancer Lett 254:178–216
Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A et al (1996) An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274:373–376
Lin E, Nemunaitis J (2004) Oncolytic viral therapies. Cancer Gene Ther 11:643–664
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM (1991) Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252:854–856
O’Shea CC (2005) DNA tumor viruses—the spies who lyse us. Curr Opin Genet Dev 15:18–26
Mastrangelo MJ, Eisenlohr LC, Gomella L, Lattime EC (2000) Poxvirus vectors: orphaned and underappreciated. J Clin Invest 105:1031–1034
Smith GL, Moss B (1983) Infectious poxvirus vectors have capacity for at least 25,000 base pairs of foreign DNA. Gene 25:21–28
Kirn DH, Wang Y, Le BF, Bell J, Thorne SH (2007) Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med 4:e353
McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, Moss B, Bartlett DL (2001) Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res 61:8751–8757
Zhang Q, Yu YA, Wang E, Chen N, Danner RL, Munson PJ, Marincola FM, Szalay AA (2007) Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res 67:10038–10046
Lee HK, Iwasaki A (2008) Autophagy and antiviral immunity. Curr Opin Immunol 20:23–29
Reading PC, Smith GL (2003) A kinetic analysis of immune mediators in the lungs of mice infected with vaccinia virus and comparison with intradermal infection. J Gen Virol 84:1973–1983
Jacobs N, Chen RA, Gubser C, Najarro P, Smith GL (2006) Intradermal immune response after infection with Vaccinia virus. J Gen Virol 87:1157–1161
Selin LK, Santolucito PA, Pinto AK, Szomolanyi-Tsuda E, Welsh RM (2001) Innate immunity to viruses: control of vaccinia virus infection by gamma delta T cells. J Immunol 166:6784–6794
Karupiah G, Blanden RV, Ramshaw IA (1990) Interferon gamma is involved in the recovery of athymic nude mice from recombinant vaccinia virus/interleukin 2 infection. J Exp Med 172:1495–1503
Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M (1993) Immune response in mice that lack the interferon-gamma receptor. Science 259:1742–1745
Smith GL, Symons JA, Khanna A, Vanderplasschen A, Alcami A (1997) Vaccinia virus immune evasion. Immunol Rev 159:137–154
Alcami A, Smith GL (1995) Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J Virol 69:4633–4639
Mossman K, Upton C, Buller RM, McFadden G (1995) Species specificity of ectromelia virus and vaccinia virus interferon-gamma binding proteins. Virology 208:762–769
Farrar MA, Schreiber RD (1993) The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol 11:571–611
Schellekens H, de Reus A, Bolhuis R, Fountoulakis M, Schein C, Ecsodi J, Nagata S, Weissmann C (1981) Comparative antiviral efficiency of leukocyte and bacterially produced human alpha-interferon in rhesus monkeys. Nature 292:775–776
Deonarain R, Alcami A, Alexiou M, Dallman MJ, Gewert DR, Porter AC (2000) Impaired antiviral response and alpha/beta interferon induction in mice lacking beta interferon. J Virol 74:3404–3409
Garcia-Sastre A, Biron CA (2006) Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882
Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP (1999) Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol 17:189–220
Biron CA, Brossay L (2001) NK cells and NKT cells in innate defense against viral infections. Curr Opin Immunol 13:458–464
Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A (2007) Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26:503–517
Martinez J, Huang X, Yang Y (2008) Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol 180:1592–1597
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991–998
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360
Dunn GP, Old LJ, Schreiber RD (2004) The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21:137–148
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7:211–217
Mantovani A, Romero P, Palucka AK, Marincola FM (2008) Tumor immunity: effector response to tumor and the influence of the microenvironment. Lancet 371:771–783
Wolfel T, Klehmann E, Muller C, Schutt KH, Meyer zum Buschenfelde KH, Knuth A (1989) Lysis of human melanoma cells by autologous cytolytic T cell clones. Identification of human histocompatibility leukocyte antigen A2 as a restriction element for three different antigens. J Exp Med 170:797–810
Marincola FM, Rivoltini L, Salgaller ML, Player M, Rosenberg SA (1996) Differential anti-MART-1/MelanA CTL activity in peripheral blood of HLA-A2 melanoma patients in comparison to healthy donors: evidence for in vivo priming by tumor cells. J Immunother 19:266–277
Fuchs EJ, Matzinger P (1996) Is cancer dangerous to the immune system? Semin Immunol 8:271–280
Aptsiauri N, Carretero R, Garcia-Lora A, Real LM, Cabrera T, Garrido F (2008) Regressing and progressing metastatic lesions: resistance to immunotherapy is predetermined by irreversible HLA class I antigen alterations. Cancer Immunol Immunother 57:1727–1733
Seliger B (2008) Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol Immunother 57:1719–1726
Menard C, Martin F, Apetoh L, Bouyer F, Ghiringhelli F (2008) Cancer chemotherapy: not only a direct cytotoxic effect, but also an adjuvant for antitumor immunity. Cancer Immunol Immunother 57:1579–1587
Ramakrishnan R, Antonia S, Gabrilovich DI (2008) Combined modality immunotherapy and chemotherapy: a new perspective. Cancer Immunol Immunother 57:1523–1529
Cancer Vaccine Fact Sheet (2008). http://www.cancer.gov/cancertopics/factsheet/cancervaccine
Kaufman HL, Taback B, Sherman W, Kim DW, Shingler WH, Moroziewicz D, DeRaffele G, Mitcham J, Carroll MW, Harrop R et al (2009) Phase II trial of Modified Vaccinia Ankara (MVA) virus expressing 5T4 and high dose Interleukin-2 (IL-2) in patients with metastatic renal cell carcinoma. J Transl Med 7:2
Stanford MM, Breitbach CJ, Bell JC, McFadden G (2008) Innate immunity, tumor microenvironment and oncolytic virus therapy: friends or foes? Curr Opin Mol Ther 10:32–37
Amato RJ (2008) Vaccine therapy for renal cancer. Expert Rev Vaccines 7:925–935
Harrop R, Drury N, Shingler W, Chikoti P, Redchenko I, Carroll MW, Kingsman SM, Naylor S, Griffiths R, Steven N et al (2008) Vaccination of colorectal cancer patients with TroVax given alongside chemotherapy (5-fluorouracil, leukovorin and irinotecan) is safe and induces potent immune responses. Cancer Immunol Immunother 57:977–986
Kaufman HL, Kim-Schulze S, Manson K, DeRaffele G, Mitcham J, Seo KS, Kim DW, Marshall J (2007) Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J Transl Med 5:60
Croft M (2003) Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev 14:265–273
Monsurro’ V, Wang E, Yamano Y, Migueles SA, Panelli MC, Smith K, Nagorsen D, Connors M, Jacobson S, Marincola FM (2004) Quiescent phenotype of tumor-specific CD8+ T cells following immunization. Blood 104:1970–1978
Marincola FM, Wang E, Herlyn M, Seliger B, Ferrone S (2003) Tumors as elusive targets of T cell-based active immunotherapy. Trends Immunol 24:335–342
Monsurro’ V, Wang E, Panelli MC, Nagorsen D, Jin P, Smith K, Ngalame Y, Even J, Marincola FM (2003) Active-specific immunization against melanoma: is the problem at the receiving end? Semin Cancer Biol 13:473–480
Wang E, Miller LD, Ohnmacht GA, Mocellin S, Petersen D, Zhao Y, Simon R, Powell JI, Asaki E, Alexander HR et al (2002) Prospective molecular profiling of subcutaneous melanoma metastases suggests classifiers of immune responsiveness. Cancer Res 62:3581–3586
Panelli MC, Wang E, Phan G, Puhlman M, Miller L, Ohnmacht GA, Klein H, Marincola FM (2002) Gene-expression profiling of the response of peripheral blood mononuclear cells and melanoma metastases to systemic IL-2 administration. Genome Biol 3:RESEARCH0035
Panelli MC, Stashower M, Slade HB, Smith K, Norwood C, Abati A, Fetsch PA, Filie A, Walters SA, Astry C et al (2006) Sequential gene profiling of basal cell carcinomas treated with Imiquimod in a placebo-controlled study defines the requirements for tissue rejection. Genome Biol 8:R8
Wang E, Worschech A, Marincola FM (2008) The immunologic constant of rejection. Trends Immunol 29:256–262
Rosenberg SA, Yang JC, Restifo NP (2004) Cancer immunotherapy: moving beyond current vaccines. Nat Med 10:909–915
Griffioen AW (2008) Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother 57:1553–1558
Hicks AM, Riedlinger G, Willingham MC, Alexander-Miller MA, von Kap-Herr C, Pettenati MJ, Sanders AM, Weir HM, Du E, Kim J et al (2006) Transferable anticancer innate immunity in spontaneous regression/complete resistance mice. Proc Natl Acad Sci USA 103:7753–7758
Shanker A, Verdeil G, Buferne M, Inderberg-Suso EM, Puthier D, Joly F, Nguyen C, Leserman L, uphan-Anezin N, Schmitt-Verhulst AM (2007) CD8 T cell help for innate antitumor immunity. J Immunol 179:6651–6662
Urosevic M, Fujii K, Calmels B, Laine E, Kobert N, Acres B, Dummer R (2007) Type I IFN innate immune response to adenovirus-mediated IFN-gamma gene transfer contributes to the regression of cutaneous lymphomas. J Clin Invest 117:2834–2846
Marleau AM, Lipton JH, Riordan NH, Ichim TE (2007) Therapeutic use of Aldara in chronic myeloid leukemia. J Transl Med 5:4
Torres A, Storey L, Anders M, Miller RL, Bulbulian BJ, Jin J, Raghavan S, Lee J, Slade HB, Birmachu W (2007) Immune-mediated changes in actinic Keratosis following topical treatment with Imiquimod 5% cream. J Transl Med 5:7
Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, Fellows-Mayle W, Storkus WJ, Walker PR, Salazar AM et al (2007) Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med 5:10
Kirn DH, Wang Y, Liang W, Contag CH, Thorne SH (2008) Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res 68:2071–2075
Worschech A, Chen N, Yu YA, Zhang Q, Pos Z, Weibel S, Raab V, Sabatino M, Monaco A, Liu H et al (2008) Systemic treatment of xenografts with vaccinia virus GLV-1h68 reveals the immunologic facts of oncolytic therapy (submitted)
Salk J (1969) Immunological paradoxes: theoretical considerations in the rejection or retention of grafts, tumors, and normal tissue. Ann NY Acad Sci 164:365–380
Rehermann B, Nascimbeni M (2005) Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol 5:215–229
Kawakami Y, Robbins P, Wang RF, Parkhurst MR, Kang X, Rosenberg SA (1998) Tumor antigens recognized by T cells. The use of melanosomal proteins in the immunotherapy of melanoma. J Immunother 21:237–246
Robbins PF, el-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, Rosenberg SA (1996) A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med 183:1185–1192
Butterfield LH, Disis ML, Fox BA, Lee PP, Khleif SN, Thurin M, Trinchieri G, Wang E, Wigginton J, Chaussabel D et al (2008) A systematic approach to biomarker discovery: preamble to “the iSBTc-FDA taskforce on Immunotherapy Biomarkers”. J Transl Med 6:81
Conflict of interest statement
This work was supported by Genelux Co.; Andrea Worschech, Dana Haddad and Aladar A Szalay have received payment or are employees of Genelux Co.
Author information
Authors and Affiliations
Corresponding authors
Additional information
A. A. Szalay and F. M. Marincola are co-senior authors.
Rights and permissions
About this article
Cite this article
Worschech, A., Haddad, D., Stroncek, D.F. et al. The immunologic aspects of poxvirus oncolytic therapy. Cancer Immunol Immunother 58, 1355–1362 (2009). https://doi.org/10.1007/s00262-009-0686-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-009-0686-7