
Abstract
Adoptive immunotherapy (AIT) using ex vivo-expanded HER-2/neu-specific T cells has shown initial promising results against disseminated tumor cells in the bone marrow. However, it has failed to promote objective responses against primary tumors. We report for the first time that alternating gamma chain cytokines (IL-2, IL-7 and IL-15) ex vivo can expand the neu-specific lymphocytes that can kill breast tumors in vitro. However, the anti-tumor efficacy of these neu-specific T cells was compromised by the increased levels of myeloid-derived suppressor cells (MDSC) during the premalignant stage in FVBN202 transgenic mouse model of breast carcinoma. Combination of AIT with the depletion of MDSC, in vivo, resulted in the regression of neu positive primary tumors. Importantly, neu-specific antibody responses were restored only when AIT was combined with the depletion of MDSC. In vitro studies determined that MDSC caused inhibition of T cell proliferation in a contact-dependent manner. Together, these results suggest that combination of AIT with depletion or inhibition of MDSC could lead to the regression of mammary tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To date, several vaccination strategies have been successfully employed to induce tumor-specific CD8+ and CD4+ T cell responses. However, such immunological responses have rarely been robust enough to achieve tumor regression [7, 48]. To overcome this obstacle by increasing the frequency of tumor-specific T cells, adoptive immunotherapy (AIT) treatments with effector cells activated and expanded ex vivo have been introduced and evaluated against a variety of cancers [8, 36]. Although AIT trials have exhibited partial effects in the case of melanoma and metastatic breast carcinoma, these therapies failed to exhibit objective responses against HER-2/neu positive breast tumors [2, 29]. Among a variety of approaches for the expansion of anti-tumor T cells, ex vivo, stimulation of T cells with bryostatin-1 (B) and ionomycin (I) has been shown to be promising in specifically activating anti-tumor effector T cells by mimicking signals through the T cell receptor, thus causing the expansion of tumor-specific effector T cells in the absence of the nominal antigen, ex vivo [6]. T cells activated in this manner and expanded in the presence of IL-2 were able to cause regression of MCA-105 pulmonary metastasis in vivo, as well as of established 4T07 mammary tumors transfected with IL-2 [5, 41]. However, IL-2 expansion may lead to the induction of regulatory T cells (T regs) and cause activation-induced cell death in T cells [20, 33, 47]. Alternatively, the gamma chain cytokines IL-7 and IL-15 are attractive candidates for AIT because of their role in the maintenance and proliferation of effector and memory T cells. IL-7 has been implicated as a pro-survival cytokine for early memory CD8+ T cell selection and maintenance, while IL-15 can protect effector CD8 + T cells from apoptosis, as well as augment their cytotoxic effects [21, 22, 30, 38, 40, 43]. Additionally, unlike IL-2, IL-7 and IL-15 do not cause activation-induced cell death, nor are they needed for the maintenance of T regs [42].
One major obstacle hindering the clinical response of cancer immunotherapy is the presence of suppressor cells. In tumor-bearing hosts, myeloid-derived suppressor cells (MDSC) have emerged as a significant mediator of immune suppression in various types of cancer. For example, MDSC accumulate in several mouse models of cancer, including the MCA-26 colon carcinoma, the 4T1 mammary carcinoma, and the neu-overexpressing mouse mammary carcinoma (MMC) [4, 19, 27]. In humans, the presence of MDSC has been associated with head and neck cancer, renal cell carcinoma, and breast cancer [10, 32, 45]. Importantly, increased levels of circulating MDSC have recently been correlated with disease stage and extensive metastatic tumor burden in patients with breast cancer [10]. It was reported that MDSC accumulate in response to tumor-derived soluble factors VEGF, GM-CSF, and M-CSF [14, 16] and inhibit anti-tumor T cell responses, possibly through the production of soluble factors nitric oxide, arginase-1, reactive oxygen species, and inhibitory cytokines such as IL-10 [3, 28, 32, 35, 46]. In particular, arginine depletion and the production of reactive oxygen species can result in downregulation of the TCR zeta chain as well as T cell arrest in the G0 phase of the cell cycle [13, 14] and impairment of IL-2 signaling [28], respectively. However, the role of MDSC in inhibiting adoptively transferred tumor-specific effector T cells has not yet been clearly defined.
The FVBN202 mouse model of spontaneously arising mammary carcinomas provides a clinically relevant model for investigating the immunotherapy of HER2/neu positive mammary tumors. These mice develop mammary carcinomas within 4-12 months of age due to the overexpression of the rat neu oncogene in their mammary glands under the regulation of the mouse mammary tumor virus (MMTV) promoter [18]. In these mice, atypical mammary hyperplasia develops prior to the occurrence of spontaneous mammary tumors and is accompanied by an increase in MDSC in the peripheral blood and spleen of FVBN202 mice during the premalignant stage, rendering animals resistant to neu-targeted immunotherapy [19, 26]. Furthermore, inoculation of MMC into FVBN202 mice causes a rapid and pronounced increase in MDSC [19]. In contrast, parental FVB mice do not express the rat neu oncogene and therefore are able to generate a robust immune response against challenge with neu-expressing MMC. These mice subsequently reject MMC, thus generating a pool of T cells with proven effectiveness against these tumors. These T cells derived from parental FVB mice are therefore ideal candidates for evaluating the efficacy of protocols for expanding neu-specific T cells in the absence of the nominal antigen, ex vivo. In addition, their FVBN202 transgenic counterpart provides an ideal model in which the effects of increased endogenous MDSC on adoptively transferred effector T cells can be evaluated. This study addresses, for the first time, the consequences of increased MDSC on the efficacy of AIT in the FVBN202 mouse model of breast carcinoma, as well as the novel finding of a restored neu-specific antibody response following the depletion of MDSC, in vivo. These findings suggest that AIT in breast cancer patients may be improved if it is combined with the inhibition or depletion of MDSC and that MDSC may also play a role in the suppression of B cell responses.
Materials and methods
Mouse model
Parental FVB (Jackson Laboratories) and FVBN202-transgenic female mice (Charles Riveer Laboratories) were used between 6 and 10 weeks of age throughout these studies. FVBN202 mice overexpress an unactivated rat neu transgene under the regulation of the MMTV promoter [18]. These mice develop premalignant mammary hyperplasia similar to ductal carcinoma in situ (DCIS) prior to the development of spontaneous carcinoma [24]. These studies have been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Commonwealth University.
Tumor cell lines
The MMC cell line was established from a spontaneous tumor harvested from an FVBN202 transgenic mouse as previously described [23]. The antigen negative variant (ANV) cell line was derived from a relapsed MMC tumor in the FVB strain as previously described and is characterized by a loss of neu expression [23]. Both cell lines were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS). Mice were challenged with 3 × 106 MMC cells i.d. where indicated.
Recombinant neu protein
The cDNAs coding for sub-domain II of the extracellular domain of rat neu (ECDII:187-332 aa) were amplified by PCR using the following primers: 5′-GTGGAATTCACCAATCGTTCCCGGGCC-3′(sense) and 5′-TCAAGCTTCACAACGCTGTGTTCC-3′ (antisense). Restriction sites are underlined. The resulting fragments were cleaved with EcoR I and Hind III and ligated into the EcoRI–Hind III fragment of pRSET A to generate the constructs. The recombinant proteins were expressed in E. coli using IPTG as an inducer of expression. Purification of His-tagged protein was performed under denaturing conditions using the Guanidinium Lysis Buffer. After elution, proteins were dialyzed in 20 mM Tris, pH 9.0 overnight (4°C). Dialyzed proteins were concentrated using 10,000 MW cut-off columns (Viva Spin), filter-sterilized and protein concentration was determined using Bradford assay.
Flow cytometry
Flow cytometry analysis was performed as previously described by our group [23]. Briefly, splenocytes were homogenized into a single cell suspension and 106 cells were aliquoted into each sample tube. Non-specific binding to Fc receptors was blocked with anti-CD16/CD32 antibody (Biolegend) for 20 min on ice. Cells were stained with surface antibodies towards various markers and incubated on ice in the dark for 20 min. Cells were washed twice and fixed with 1% paraformaldehyde or were washed again in 1X Annexin V buffer (BD Pharmingen) and the Annexin V staining protocol was followed. Samples were run on a Beckman Coulter FC 500 and analyzed using Summit version 4.3 software.
Cytotoxicity assay
Neu-specific effector lymphocytes were cultured with MMC at 10:1 and 20:1 E:T ratios in complete medium (RPMI-1640 supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FBS) and 20 U/mL recombinant IL-2 (Peprotech) in six well culture dishes. After 24 h, 3 mL fresh media was added to the existing 3 mL of media. After 48 h, cells were harvested and stained for neu (anti-c-ErbB2/c-Neu, Calbiochem), Annexin V and PI according to the manufacturer’s protocol (BD Pharmingen). Flow cytometry was used to analyze the viability of neu positive cells.
IFN-γ ELISA
Neu-specific effector lymphocytes were cultured in complete medium at a 10:1 ratio with irradiated MMC cells or ANV cells (15,000 rad) for 24 h. Supernatants were collected and stored at −80°C until used. IFN-γ was detected using a Mouse IFN-γ ELISA Set (BD Pharmingen) according to the manufacturer’s protocol. Results are reported as the mean values of duplicate wells.
Expansion of effector T cells from FVB mice
FVB parental mice were inoculated with 5 × 106 MMC cells and splenocytes harvested after 20–25 days. Splenocytes (106 cells/mL) were stimulated in complete medium containing 15% FBS with bryostatin-1 (5 nM) and ionomycin (1 μM) along with 80 U/mL of IL-2 (Peprotech) for 16 h. Cells were washed three times and cultured at 106 cells/mL in complete medium with 40 U/mL IL-2 and media was changed every other day for a total of 7 days. Cells expanded with alternating gamma chain cytokines were cultured on day 1 with 10 ng/mL IL-7 and 10 ng/mL IL-15 (Peprotech). On day 2, 40 U/mL of IL-2 was added. Medium was changed on days 3 and 5, each time culturing with 10 ng/mL each of IL-7 and IL-15 and injections were done on day 7. Aliquots of cells were taken at indicated time points and samples were stained with combinations of the following antibodies and assessed by flow cytometry: FITC-CD25, FITC-CD62L, FITC-CD8, PE-CD44, PE-CD69, PE-CD8, PE/Cy5-CD4, PE/Cy5-CD8, PE/Cy5-CD69 from Biolegend and PE-CD25, PE-CD4 from BD Pharmingen. All antibodies were used at the manufacturer’s recommended concentration.
Adoptive immunotherapy
Twenty-four hours prior to AIT, FVBN202 mice were treated with Cyclophosphamide (CYP, 100 mg/kg) by i.p. injection in order to induce lymphopenia. Mice were challenged i.d. with 3 × 106 MMC cells and then received 70 × 106 T cells by tail vein injection later the same day. Tumor growth was monitored by digital caliper and tumor volumes were calculated by: V(volume) = [L(length) × W(width)2]/2. Blood was collected from the orbital sinus periodically to determine antibody responses and levels of CD11b + Gr1+ cells by flow cytometry. At the termination of the experiment, splenocytes were harvested and stained using the same antibodies as indicated above.
Depletion of MDSC in vivo
Monoclonal antibody against the surface antigen Gr1 was purified from the RB6-8C5 hybridoma by collecting supernatant from the CELLine CL 1,000 flask (IBS Integra Biosciences) according to the user manual. Supernatant was stored at −80°C until IgG purification using a MEP-Hypercell column. Where indicated, mice were injected i.p. with 250 μg of anti-Gr1 antibody for a total of 6 times at 3-day intervals, followed by a final injection of 200 μg on day 25. Depletion was verified by flow cytometry of the peripheral blood for the CD11b and Gr1 surface markers. The dose of anti-Gr1 antibody was proven completely effective in mice bearing MMC tumors that were 25 mm3 or smaller.
Isolation of MDSC in vitro
Gr1+ cells were isolated using an EasySep PE Selection kit from StemCell Technologies. The protocol from the manufacturer was followed using splenocytes homogenized from MMC tumor-bearing FVBN202 mice labeled with 2 μg/mL of PE-Gr1 or PE-CD11b antibody from Biolegend. Purity of Gr1 + cells was confirmed by flow cytometry and was >90%.
In vitro T cell proliferation and BrdU labeling
T cell stimulations were done in 96 well plates. Plates were coated with 10 μg/mL of anti-CD3 (BD Pharmingen) and were washed three times with PBS after 24 h to remove any unbound antibody. Splenocytes (106 cells/mL in complete media), were labeled by adding 10 μM BrdU (BD Pharmingen) directly to the culture medium. Soluble anti-CD28 antibody (BD Pharmingen) was also added to the culture medium at 1 μg/mL. Cells were plated at 2 × 105 cells/well and were allowed to proliferate for 72 h at 37°C, 5% CO2. Staining for BrdU was done following the protocol from the manufacturer (BD Pharmingen) using the FITC-conjugated anti-BrdU flow kit. Where indicated, MDSC were depleted from the splenocyte populations using the PE Selection protocol above with either PE-Gr1 or PE-CD11b antibodies (Biolegend). Isolated MDSC were added to wells where indicated at a 1:2 MDSC to splenocyte ratio either in the absence of a transwell insert, or in the top chamber of a transwell insert with 8.0 μm pore (Corning Life Sciences).
ELISA
Blood was collected from mice via the retro-orbital sinus, allowed to sit at room temperature for 10 min, and then spun for 10 min at 10,000 rpm. Serum was harvested and stored at −80°C until used. For measuring the antibody response against neu, 96 well plates were coated with 10 μg/mL of the ECDII and incubated overnight at 4°C. Plates were washed with PBS + 0.05% Tween-20 and blocked with 2% skim milk for 1 h. After washing, fivefold serial dilutions of the sera were added (100 μL/well) and incubated for 2 h at room temperature. Horse-radish-peroxidase (HRP)-conjugated anti-mouse IgG1 from Caltag was added at a 1:2,000 dilution for 1 h. Plates were washed and reactions developed by adding 100 μL/well of the TMB Microwell peroxidase substrate (Kierkegaard and Perry). The reaction was stopped with 2 M H2SO4, and the OD read at 450 nm. Mean antibody titers were then calculated.
Results
Bryostatin-1/Ionomycin (B/I) stimulation followed by IL-2 expansion generates highly activated neu-specific effector T cells
Since parental FVB mice recognize the rat neu protein as a foreign antigen and are subsequently able to reject MMC, whereas FVBN202 mice often tolerate neu protein and are unable to reject MMC, FVB mice were used as donors for AIT transfers into FVBN202 recipients in these studies. Since B/I selectively activates effector/memory T cells regardless of their antigen specificity [41], we sensitized FVB mice with MMC cells in order to increase the pool of neu-specific effector/memory T cells prior to T cell harvest for B/I activation ex vivo. We first compared the populations of CD4+ and CD8+ T cells from these donors immediately after harvest, after activation with B/I, and after a 7-day expansion with IL-2. Representative data from duplicate experiments are presented in Fig. 1a. FVB donor splenocytes contained 35 and 9% CD4+ and CD8+ T cells, respectively. These populations were similar immediately after B/I activation (27 and 12% CD4+ and CD8+, respectively) but were greatly increased after 7 days of culture with IL-2 (55 and 32% CD4+ and CD8+, respectively). Furthermore, IL-2 treatment increased the absolute number of viable T cells by 9.5-fold over the cell number that was cultured after B/I expansion (Fig. 1b). Annexin V+ CD4+ populations remained nearly constant during ex vivo expansion, starting at 20% on day 0 compared with 24% after B/I activation and cytokine treatment (Fig. 1c). The CD8+ T cells, however, showed a marked increase in Annexin V+ staining from day 0 to 7, with the fresh splenocytes being only 13% Annexin V+, while the post-B/I and post-cytokine values were 23 and 54%, respectively (Fig. 1c).

Phenotype analysis of neu-specific anti-tumor T cells before and after activation with B/I and expansion with IL-2. a Flow cytometry analysis of total CD4+ and CD8+ populations in the lymphocyte region prior to B/I activation (pre-B/I), 16 h after B/I activation (post-B/I), and 7 days after expansion in the presence of IL-2 (post-IL-2). Representative data from two independent experiments are shown. b Absolute numbers of viable cells were determined from three independent experiments before and after expansion with IL-2 as determined by trypan blue exclusion using a hemocytometer. c Viability of T cell subsets as determined by Annexin V negative population within gated CD4+ or CD8+ lymphocyte regions. d–f Flow cytometry analysis performed on gated CD4+ or CD8+ T cells to determine percentage of memory T cells (CD44+ CD62L+), effector T cells (CD44+ CD62L−), early effector T cells (CD25+) and very early effector T cells (CD69+)
In order to determine T cell phenotypes, flow cytometry was performed for memory T cells (CD44+ CD62L+) and effector T cells (CD44+ CD62L−), as well as for the activation marker CD25, and the very early activation marker CD69, in both the CD4+ and CD8+ populations (Fig. 1d). As expected, B/I activation greatly increased the effector phenotype in the CD4+ population from 16 to 79% and in the CD8+ population from 5 to 66% while naïve (CD44− CD62L+) and memory (CD44+ CD62L+) CD4+ and CD8+ T cells were greatly decreased (Fig. 1d). After IL-2-induced expansion, both CD4+ and CD8+ T cells showed a marked increase in CD44+ CD62L+ memory T cells (74 and 58% in the CD4+ and CD8+ compartments, respectively), while maintaining increased levels of CD44+ CD62L− effector T cells compared to pre-B/I treatment (16 vs. 22% and 5 vs. 38%, respectively). Additionally, the expression of CD25 on CD4+ and CD8+ T cells was markedly increased to over 80% in both cases after B/I activation, and increased to over 90% in both populations after IL-2 expansion (Fig. 1e). The very early activation marker, CD69, was greatly increased from about 0.1–0.2% in fresh splenocytes to 87% in CD4+ T cells and 93% in CD8+ T cells after B/I activation. This value, however, dropped again after a 7-day culture with IL-2, with 0.8% CD69 expression in CD4+ T cells, and, notably, 3% remaining in the CD8+ T cells. However, most cells retained a late effector phenotype (CD44+ CD69−) on day 7 (Fig. 1f).
To confirm anti-tumor efficacy in vitro, T cells derived from MMC sensitized FVB mice prior to (Pre-B/I) or after a 7-day ex vivo expansion (post-cytokine) were co-cultured with MMC target cells (E:T ratio of 10:1) for 48 h followed by staining with antibodies directed towards neu, Annexin V, and PI. Control wells were seeded with MMC in the absence of T cells (Medium) (Fig. 2a). Gating on neu positive cells and analyzing the percentages of Annexin V and PI positive cells allowed for the determination of specific killing of the neu positive MMC cells by T cells. Viability of MMC in the absence of T cells was 86% (Annexin V and PI negative) while it dropped to 44% in the presence of the freshly isolated T cells (Pre-B/I). Viability of MMC was further decreased to 27% when cultured with B/I-activated, IL-2-expanded T cells (post-cytokine) (Fig. 2a) Absolute numbers of viable MMC also reflect increased anti-tumor efficacy of B/I-activated, IL-2-expanded T cells compared to freshly isolated T cells (P = 0.026) (Fig. 2b). No killing was detected against the neu negative tumor variant, as determined by trypan blue exclusion (data not shown). Increased anti-tumor efficacy of the B/I-activated, IL-2-expanded T cells was due to an increased frequency of the neu-specific T cells compared to the freshly isolated T cells, as determined by IFN-γ ELISA (data not shown).

Anti-tumor efficacy of neu-specific effector T cells before and after activation with B/I and expansion with IL-2. a Annexin V and PI analyses of gated neu positive MMC after 48 h of culture alone (top), or with neu-specific lymphocytes before (middle) or after (bottom) B/I activation and 7 days expansion in the presence of IL-2. Representative data of gated neu positive cells are shown from two independent experiments. b Cell counts using trypan blue exclusion for quantification of the total number of viable MMC. Data are averages of 2–4 experiments ± SEM
T cell expansion using an alternating gamma chain cytokine regimen increases T cell expansion and viability as well as anti-tumor efficacy in vitro
In order to select the best method of T cell expansion for AIT we sought to determine if expansion of T cells in the presence of alternating gamma chain cytokines may have advantages over expansion in the presence of IL-2 alone in terms of the expansion rates, viability, phenotype, and anti-tumor efficacy in vitro. Therefore, we expanded T cells using alternating gamma chain cytokines, i.e., adding a combination of IL-7 and IL-15 (10 ng/mL) on days 1, 3, and 5 with a one-time “pulse” of IL-2 (40 U/mL) on the second day. This type of expansion increased the percentage of CD8+ T cells (32% in Fig. 1a vs. 47% in Fig. 3a) and showed an 11-fold expansion in overall viable T cell number as compared to a 9.5-fold expansion with IL-2 treatment (Fig. 1b vs. 3b). Alternating gamma chain cytokines also greatly enhanced the viability of T cells on day 7 of culture when compared to expansion with IL-2 alone (Fig. 3c shows 13% Annexin V+ CD4+ T cells and 11% Annexin V+ CD8+ T cells compared with 24% Annexin V+ CD4+ T cells and 54% Annexin V+ CD8+ T cells in Fig. 1c). Of note, there were more CD44+ CD62L− effector T cells in both the CD4+ and CD8+ compartments after expansion using alternating gamma chain cytokines as compared to expansion with IL-2 alone (33 vs. 22% CD4+ T cells and 48 vs. 38% CD8+ T cells, Figs. 1d, 3d). While the percentage of CD4+ CD25+ T cells and CD4+ CD69+ T cells remained unchanged, CD8+ CD25+ T cells decreased from 92 to 69% and CD8+ CD69+ T cells decreased from 3 to 0.3% in comparing the IL-2-expanded T cells with alternating cytokine-expanded T cells, respectively (Figs. 1d, 3d). The cytotoxic effect of T cells against MMC, in vitro, was also greater using alternating gamma chain cytokines compared to that using IL-2 (27% viable MMC in Fig. 2a vs. 14% viable MMC in Fig. 3e). Absolute numbers of viable MMC also reflects a slight increase in anti-tumor efficacy of B/I-activated, alternating cytokine-expanded T cells compared to IL-2-expanded T cells (0.5 × 106 in Fig. 2b vs. 0.4 × 106 in Fig. 3e). Cells expanded in alternating cytokines also exhibited a strong IFN-γ response when stimulated with irradiated neu positive MMC (15,000 rad), but not with neu-negative ANV cells (P = 0.006), thus confirming the neu specificity of these cells (Fig. 3f).

Characterization of neu-specific anti-tumor effector T cells expanded ex vivo using “alternating” gamma chain cytokines. a Flow cytometry analysis was performed to determine the percentage of CD4+ and CD8+ T cells after B/I activation and 7 days expansion with alternating gamma chain cytokines. Representative data from three experiments are shown. b Absolute numbers of lymphocytes were determined by trypan blue exclusion cell counts using a hemocytometer. Data are mean of three experiments ± SD. c Viability of T cell subsets as determined by Annexin V negative population within gated CD4+ or CD8+ lymphocyte regions. d Flow cytometry analysis was performed on gated CD4+ or CD8+ T cells to determine the percentage of memory T cells (CD44+ CD62L+), effector T cells (CD44+ CD62L−), early effector T cells (CD25+) and very early effector T cells (CD69+) after B/I activation and 7 days expansion with alternating cytokines. e MMC cells were cultured for 48 h with medium (MMC) or in the presence of the neu-specific lymphocytes activated with B/I and expanded with alternating gamma chain cytokines (MMC+ T cells). Annexin V and PI analyses were performed on gated neu positive MMC. Cell counts using trypan blue exclusion were done for quantification of the total number of viable MMC. f IFN-γ secretion by T cells in the presence or absence of irradiated neu + MMC or neu-ANV. Results are the average of duplicates ± SD
Adoptive transfer of T cell subsets expanded ex vivo with alternating gamma chain cytokines inhibits tumor growth when combined with the depletion of MDSC in vivo
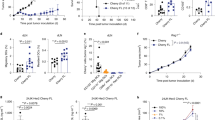
We hypothesized that an immunosuppressive environment characterized by a drastic increase in CD11b+ Gr1+ MDSC in the peripheral blood and spleens of FVBN202 mice would inhibit the anti-tumor efficacy of neu-specific T cells prepared from MMC sensitized FVB donor mice. To test this hypothesis, FVB mice were inoculated with MMC (5 × 106 cells/mouse) and donor splenocytes were prepared 21 days after tumor challenge, when animals had completely rejected MMC. T cells were activated with B/I and were then expanded in the presence of alternating gamma chain cytokines as described above (Fig. 3). All FVBN202 mice were treated with i.p. injection of CYP (100 mg/kg) in order to create lymphopenia. Flow cytometry of the peripheral blood before, and 24 h after, CYP injection, showed no effect of this drug on the CD11b+ Gr1+ population. FVBN202 mice were challenged with MMC (3 × 106) 24 h after CYP treatment. MMC-challenged mice then received no treatment, the alternating gamma chain cytokine-expanded T cells alone (i.v. injection of 70 × 106 lymphocytes/mouse), or AIT combined with the depletion of MDSC by i.p. injection of an anti-Gr1 antibody starting 6 days after tumor inoculation and continuing every 3 days for a total of six injections, followed by a final injection of 200 μg on day 25 (Fig. 4a). AIT alone offered no protection against MMC (Fig. 4a). However, the in vivo depletion of MDSC improved the efficacy of AIT and caused significant tumor inhibition (P = 0.001 for week 4 and P = 0.0003 for week 5). The efficacy of MDSC depletion was above 98% 9 days after tumor challenge (data not shown). Flow cytometry analysis of blood collected from each group 24 days after the tumor challenge showed that the group receiving Gr1 depletions had a significantly reduced percentage of MDSC (Fig. 4b). However, it is noteworthy that 38% of the granulocytes were still CD11b + Gr1 + at this time, a problem that we believe to be caused by slightly increased tumor burden in these mice leading to increased recruitment of MDSC (Fig. 4b). Our preliminary studies showed that the optimal dose of anti-Gr1 antibody was effective in depleting MDSC, however, we were not able to increase the frequency of antibody injections because of toxicity of the antibody (data not shown). Flow cytometry analysis of MDSC levels in the blood on day 35 (10 days after the last injection of anti-Gr1 antibody) showed that levels of MDSC were fully replenished in the Gr1 depletion group (data not shown). AIT using either CD4 + or CD8+ T cells alone, as well as the administration of anti-Gr1 antibody alone, did not cause tumor inhibition (data not shown).

AIT after expansion with “alternating” gamma chain cytokines combined with the depletion of MDSC in vivo can result in tumor inhibition and restoration of the neu-specific antibody response. a Tumor growth measurements of mice that received CYP followed by MMC challenge (3 × 105) were given no treatment (circles), 70 × 106 adoptively transferred T cells (triangles), or 70 × 106 adoptively transferred T cells followed by administration of 250 μg of anti-Gr1 antibody on days 6, 9, 12, 15, 18, and 21 and 200 μg on day 25 after tumor innoculation for depletion of MDSC (squares). Data points represent the averaged tumor volumes of 4–6 mice per group. b Efficacy of MDSC depletions as measured by flow cytometry analysis of blood using antibodies against CD11b and Gr1. Values are the average percentage of CD11b+ Gr1+ cells from the granulocyte regions of 4–6 mice per group 24–30 days after tumor inoculation ± SD. c Mean IgG1 antibody titers against ECDII in serum collected from animals (n = 4) 21 days after MMC inoculation
Since the ECD-specific antibody response is also involved in the protection against neu positive mammary tumors [11, 12, 25], we sought to determine whether FVBN202 mice mounted antibody responses against the ECD following AIT. Serum taken from mice receiving adoptive transfer of T cells expanded with alternating gamma chain cytokines with and without the in vivo depletion of MDSC, along with control mice, indicated that only mice that were depleted of Gr1+ cells were able to mount an antibody response against the ECD (P = 0.006) (Fig. 4c).
The presence of MDSC inhibits CD3/CD28-induced proliferation of T cells in vitro in a contact-dependent manner
To determine the mechanisms by which MDSC suppress anti-tumor immune responses, splenocytes were isolated from MMC tumor-bearing and tumor-free FVBN202 mice. The tumor-bearing mice had a large influx in MDSC in their spleens as compared to tumor-free mice [19]. Splenocytes were labeled with the thymidine analog BrdU and stimulated with antibodies against CD3 (10 μg/mL, plate-bound) and CD28 (1 μg/mL, soluble), or were left unstimulated to serve as a control. After 3 days, cells were stained using anti-CD4 and anti-CD8 antibodies and analyzed for BrdU uptake by flow cytometry. We found higher proliferation of CD4+ T cells from tumor-free mice compared to those from tumor-bearing animals (Fig. 5a, 91 vs. 59%, P = 0.002). The same trend was seen for CD8+ T cells (Fig. 5a, 93 vs. 70%). Similar trends were detected while analyzing absolute numbers of CD4+ and CD8+ T cells (Fig. 5b). Fig. 5b shows that the average number of BrdU+ CD4+ and BrdU+ CD8+ T cells from tumor-free mice is 3.3-fold higher than that of BrdU+ CD4+ and BrdU+ CD8+ T cells from tumor-bearing mice (P = 0.003 for CD4+ and P = 0.0004 for CD8+ T cells). Using lymphocytes derived from FVB donors we also found similar patterns of the MDSC-mediated suppression of T cell proliferation in vitro (data not shown). To confirm that the suppression of T cell proliferation in FVBN202 splenocytes was caused by the presence of elevated MDSC, we depleted MDSC in vitro, from the splenocytes of the tumor-bearing animals. As seen in Fig. 5a, the depletion of MDSC (tumor-bearing-MDSC) significantly restored the proliferative responses of both CD4+ (87% BrdU+) and CD8+ (92% BrdU+) T cells over those seen in total splenocytes (P = 0.028 for CD4+ and P = 0.009 for CD8+ T cells). Similar trends were found while comparing the absolute numbers of CD4+ and CD8+ T cells, showing a 3.1-fold increase in proliferation in the CD4+ population (P = 0.027) and 2.5-fold increase in the CD8+ population (P = 0.012) over total splenocytes from tumor-bearing mice (Fig. 5b). To determine if the anti-proliferative effects of MDSC on T cells were contact-dependent, CD11b+ cells were depleted from the splenocytes of tumor-bearing and tumor-free mice and equal numbers of lymphocytes (5 × 105) were stimulated in the lower chamber of a plate containing a Transwell insert. CD11b+ cells depleted from tumor-bearing mice were then added to the lower chamber of the plate, or were added to the top chamber of the Transwell insert, where they were separated from the lymphocytes by an 8.0 υm pore. Staining for BrdU incorporation on day 3 showed potent inhibition when CD11b+ cells were added back to the cultures, but only in the absence of a Transwell insert (Fig. 5c). There was a total lack of inhibition observed when CD11b+ cells were separated from the T cells by a transwell insert (Fig. 5c). Similar trends were detected while analyzing the absolute numbers of CD4+ and CD8+ T cells (data not shown).

MDSC-mediated inhibition of T cell proliferation in a contact-dependent manner. a–b Percentage and absolute numbers of CD4+ and CD8+ cells that were positive for BrdU incorporation after a 3-day culture with anti-CD3 and anti-CD28 antibodies using total splenocytes from tumor-free or tumor-bearing FVBN202 mice with and without MDSC depletion in vitro. Data are averages of two separate experiments ± SD. c Splenocytes from FVBN202 mice were depleted of CD11b+ cells, and added to the bottom of a transwell culture dish and pulsed with BrdU. Where indicated, MDSC from the tumor-bearing FVBN202 mice were added, either directly to the splenocytes (Contact) or in the top chamber of a transwell insert (No contact). Cells were stimulated with anti-CD3 and anti-CD28 antibodies for 3 days and analyzed for BrdU incorporation in the CD4+ and CD8+ populations by flow cytometry as before. Data are presented from duplicate experiments
Discussion
The adoptive transfer of tumor-specific T cells that have been activated and expanded in vitro is a promising means of systemically treating residual cancers after resection of the primary tumor. The presence of immune suppressor cells has, however, become a substantial obstacle to the success of these treatments. In particular, MDSC represent a potent population of suppressor cells that are elevated in many different types of cancer, including neu positive mammary tumors in FVBN202 mice, and have been associated with suppression of T cell responses by multiple mechanisms. T regs were also reported to suppress anti-tumor immune responses. We looked at T regs in the blood, spleen, lymph nodes and the tumor site of FVBN202 mice and found no changes in the number of CD4+ CD25+ Foxp3+ T regs (data not shown). Since we wanted to determine whether MDSC could suppress robust T cell responses even against allogeneic antigen, we used FVB mice as donors of T cells. Although using such an allogeneic system will reduce clinical application of the proposed AIT regimen, failure of such effector T cells in the rejection of MMC tumor cells attests to the strength of in vivo suppression by MDSC, and upon proving that MDSC depletion does indeed facilitate an otherwise ineffective AIT, we would next hope to use these procedures with the expansion of splenocytes from tumor sensitized FVBN202 mice. We also showed for the first time that alternating gamma chain cytokine conditions are more effective than IL-2 alone for the expansion of neu-specific anti-tumor effector T cells. Having compared IL-2 with alternating gamma chain cytokines (IL-7 and IL-15 with a one-time dose of IL-2), we found that the latter was superior to the former in expanding T cells with a higher overall viability, as well as a higher proportion of effector T cell phenotypes, which could readily exhibit anti-tumor activity. In fact, alternating gamma chain cytokine-expanded T cells showed a higher anti-tumor efficacy than IL-2-expanded T cells, as evaluated by cytotoxicity assay in vitro. However, adoptive transfer of such effector T cells did not overcome the pre-existing immune suppressive microenvironment in FVBN202 animals. We have previously shown that these mice have increased levels of MDSC, even at the premalignant stage due to mammary hyperplasia [19, 24]. Interestingly, the combination of depleting MDSC in vivo with AIT using neu-specific T cells resulted in a robust tumor regression following AIT. Complete rejection of MMC tumors by the effector T cells, however, was hindered by the fact that antibody-mediated MDSC depletion in FVBN202 mice was incomplete. This was due to increasing numbers of MDSC in tumor-bearing mice over time following the cessation of injection of anti-Gr1 antibody, as preliminary depletions (9 days after tumor challenge) were nearly 100% effective (data not shown). To achieve MDSC depletion, we were not able to inject the rat anti-mouse Gr1 antibody more than six times because of its toxicity. Because of such limitations in the control of MDSC in vivo, we were able to perform prophylactic studies only. Therapeutic efficacy of this strategy on established tumors remains to be determined using alternative drugs such as gemcitabine for selective elimination of MDSC [26]. Sporadic spontaneous tumor development after a long latency in FVBN202 mice makes it difficult to test whether AIT combined with the inhibition of MDSC could also protect animals against spontaneous tumor development.
Although other groups have reported the role of MDSC in suppression of anti-tumor T cell responses [3, 15, 16], we report for the first time that MDSC also suppress humoral immune responses following AIT so that depletion of MDSC in vivo restored anti-neu antibody responses in FVBN202 mice. This is very important because it has been reported that collaboration of humoral and cellular immune responses is required for optimal elimination of HER-2/neu positive tumors [2]. In addition, a novel HER-2/neu-specific antibody, Pertuzumab, is currently in phase III trial and has been shown to have anti-tumor function through recognizing and blocking the dimerization domain of HER-2/neu, ECDII [1, 37]. Curiously, there was no antibody response in the groups that received AIT alone, indicating that the presence of adoptively transferred CD4+ T cells alone is not sufficient to facilitate IgG1 isotype switching by the recipients’ B cells because of the presence of MDSC. It is unclear at this point whether the restoration of the antibody response results from lifting MDSC suppression of adoptively transferred helper T cells, or whether MDSC directly suppress B cell responses. We performed multiplex cytokine array analysis of the sera collected from the experimental groups above following AIT but did not detect any changes in the level of IL-4 (unpublished observation). The role of MDSC in the suppression of the humoral response should be further investigated and could have many applications beyond cancer immunotherapy, since increased MDSC have also been seen in some parasitic infections such as Trypanosoma cruzi [17] and in cases of polymicrobial sepsis [9].
In order to further confirm that failure of AIT to induce regression of MMC in FVBN202 mice was indeed due to inhibition of T cell function by MDSC, we performed in vitro assays to assess MDSC-mediated inhibition of T cell proliferation. Consistent with the fact that tumor-bearing animals exhibit about a fourfold increase of MDSC in the granulocyte region of their splenocytes [19], total splenocytes from tumor-bearing mice showed a marked reduction in the number and percentage of proliferating T cells. Significantly, depleting MDSC from the culture using either an anti-CD11b or an anti-Gr1 antibody caused the restoration of TCR-mediated T cell proliferation. These observations suggest that MDSC suppress TCR-induced proliferation of T cells.
The possible contact-dependent mechanism of T cell suppression by MDSC has been an area of some debate. Using different tumor models, most groups have found suppression of T cells by MDSC to be mediated by soluble factors such as arginase-1, nitric oxide, reactive oxygen species and peroxynitrites [34, 39]. In particular, arginase-1 has been implicated in downregulation of the TCR zeta chain and the induction of this enzyme has been linked to tumor-derived soluble factors such as prostaglandin E2 as well as IL-4 and IL-13 [13, 34, 35]. Furthermore, arginine-depleted conditions can cause TCR zeta downregulation in the absence of cell-to-cell contact [35]. We were therefore surprised to see that in our model, cell-to-cell contact was required for the suppression of T cell proliferation. Although other mechanisms may be involved in suppression of cytotoxic responses by T cells, we report here that no suppression of T cell proliferation was seen when MDSC were added to the top chamber of a transwell insert. Nagaraj et al. [31] have recently shown that direct cell-to-cell contact between CD8+ T cells and MDSC causes nitration of tyrosines in the TCR–CD8 complex, therefore disrupting binding of specific antigen-MHC class I complexes to the TCR’s of OT-1 transgenic T cells. It is unclear, however, if the effects of this nitration may be exaggerated due to transgenic expression of the TCR and what role this mechanism may play in a TCR non-transgenic model. Furthermore, Gabrilovich et al. [15] have reported that blocking the MHC class I molecules expressed on the surface of MDSC can reverse suppression of CD8+ T cells, which involved MDSC production of nitric oxide, but reported that these cells did not suppress CD4+ T cells responses towards MHC class II presented peptides. In 2000, Kusmartsev et al. showed inhibition of CD3/CD28 T cell activation by MDSC isolated from mice bearing MCA-26 colon carcinomas. However, this suppression was reversed by the addition of a superoxide dismutase mimetic and a nitric oxide synthase inhibitor and a possible role of contact was not investigated, and proliferation was not determined in separate populations of CD4+ and CD8+ T cells [27]. In contrast, we show here that MDSC from mice bearing HER2/neu + mammary carcinomas inhibit the proliferation of both CD4+ and CD8+ T cells in contact-dependent manner. Therefore, although contact between MDSC and CD8+ T cells has been speculated to be important in inhibiting the IFN-γ response of CD8+ T cells towards specific peptide, we show here that contact is also necessary to inhibit CD3/CD28 T cell stimulation and can affect the proliferation of both the CD4+ and CD8+ T cells populations.
Furthermore, we have shown that endogenous MDSC inhibit T cell proliferation in the splenocytes of tumor-bearing FVBVN202 mice. Therefore, it is likely that elevated levels of MDSC during the premalignant stage may generate an immunosuppressive microenvironment in these animals that could inhibit anti-tumor efficacy of AIT. We have previously shown that increased MDSC in FVBN202 mice during the premalignant stage was associated with mammary gland hyperplasia, and this was correlated with the failure of pre-existing neu-specific immune responses to prevent spontaneous mammary carcinomas in these mice [19, 24]. We have also reported the existence of an immune suppressive microenvironment at the tumor lesions of FVBN202 mice, as evidenced by increased levels of IL-10, IL-10 receptor, SOCS-1, and SOCS-3 [44]. Such a microenvironment could then inhibit AIT unless it is combined with the depletion of MDSC, suggesting that MDSC may be the key cells initiating the cascade of events leading to tumor-specific immune suppression.
References
Allen SD, Garrett JT, Rawale SV, Jones AL, Phillips G, Forni G, Morris JC, Oshima RG, Kaumaya PT (2007) Peptide vaccines of the HER-2/neu dimerization loop are effective in inhibiting mammary tumor growth in vivo. J Immunol 179(1):472–482
Bernhard H, Neudorfer J, Gebhard K, Conrad H, Hermann C, Nährig J, Fend F, Weber W, Busch DH, Peschel C (2008) Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol Immunother 57(2):271–280
Bronte V, Zanovello P (2005) Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol 5(8):641–654
Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2006) Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176(1):284–290
Chin CS, Graham LJ, Hamad GG, George KR, Bear HD (2001) Bryostatin/ionomycin-activated T cells mediate regression of established tumors. J Surg Res 98(2):108–115
Chin CS, Miller CH, Graham L, Parviz M, Zacur S, Patel B, Duong A, Bear HD (2004) Bryostatin 1/ionomycin (B/I) ex vivo stimulation preferentially activates L-selectinlow tumor-sensitized lymphocytes. Int Immunol 16(9):1283–1294
Curigliano G, Spitaleri G, Pietri E, Rescigno M, De Braud F, Cardillo A, Munzone E, Rocca A, Bonizzi G, Brichard V, Orlando L, Goldhirsch A (2006) Breast cancer vaccines: a clinical reality or fairy tale? Ann Oncol 17(5):750–762
Dang Y, Knutson KL, Goodell V, Goodell V, dela Rosa C, Salazar LG, Higgins D, Childs J, Disis ML (2007) Tumor antigen-specific T-cell expansion is greatly facilitated by in vivo priming. Clin Cancer Res 13(6):1883–1891
Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL (2007) MyD88-dependent expansion of an immature GR-1(+) CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med 204(6):1463–1474
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ (2008) Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 57(2):271–280
Dela Cruz JS, Lau SY, Ramirez EM, De Giovanni C, Forni G, Morrison SL, Penichet ML (2003) Protein vaccination with the HER2/neu extracellular domain plus anti-HER2/neu antibody-cytokine fusion proteins induces a protective anti-HER2/neu immune response in mice. Vaccine 21(13–14):1317–1326
Emens LA, Reilly RT, Jaffee EM (2005) Breast cancer vaccines: maximizing cancer treatment by tapping into host immunity. Endocr Relat Cancer 12(1):1–17
Ezernitchi AV, Vaknin I, Cohen-Daniel L, Levy O, Manaster E, Halabi A, Pikarsky E, Shapira L, Baniyash M (2006) TCR zeta down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J Immunol 177(7):4763–4772
Gabrilovich D (2004) Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol 4(12):941–952
Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM (2001) Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol 166(9):5398–5406
Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borello I, Zanovello P, Bicciato S, Bronte V (2006) Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 116(10):2777–2790
Goni O, Alcaide P, Fresno M (2002) Immunosuppression during acute Trypanosoma cruzi infection: involvement of Ly6G (Gr1(+))CD11b(+)immature myeloid suppressor cells. Int Immunol 14(10):1125–1134
Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ (1992) Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA 89(22):10578–10582
Habibi M, Kmieciak M, Graham L, Morales JK, Bear HD, Manjili MH (2008) Radiofrequency thermal ablation of breast tumors combined with intralesional administration of IL-7 and IL-15 augments anti-tumor immune responses and inhibits tumor development and metastasis. Breast Cancer Res Treat
Haux J, Johnsen AC, Steinkjer B, Egeberg K, Sundan A, Espevik T (1999) The role of interleukin-2 in regulating the sensitivity of natural killer cells for Fas-mediated apoptosis. Cancer Immunol Immunother 48(2–3):139–146
Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, Busch DH (2004) Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8 + memory T cell subsets. Proc Natl Acad Sci USA 101(15):5610–5615
Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R (2003) Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4(12):1191–1198
Kmieciak M, Knutson KL, Dumur CI, Manjili MH (2007) HER-2/neu antigen loss and relapse of mammary carcinoma are actively induced by T cell-mediated anti-tumor immune responses. Eur J Immunol 37(3):675–685
Kmieciak M, Morales JK, Morales J, Bolesta E, Grimes M, Manjili MH (2008) Danger signals and nonself entity of tumor antigen are both required for eliciting effective immune responses against HER-2/neu positive mammary carcinoma: implications for vaccine design. Cancer Immunol Immunother 57(9):1391–1398
Knutson KL, Almand B, Dang Y, Disis ML (2004) Neu antigen-negative variants can be generated after neu-specific antibody therapy in neu transgenic mice. Cancer Res 64(3):1146–1151
Ko HJ, Kim YJ, Kim YS, Chang WS, Ko SY, Chang SY, Sakaguchi S, Kang CY (2007) A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res 67(15):7477–7486
Kusmartsev SA, Li Y, Chen SH (2000) Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol 165(2):779–785
Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM (2002) Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 168(2):689–695
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314(5796):126–129
Mueller YM, Makar V, Bojczuk PM, Witek J, Katsikis PD (2003) IL-15 enhances the function and inhibits CD95/Fas-induced apoptosis of human CD4+ and CD8+ effector-memory T cells. Int Immunol 15(1):49–58
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI (2007) Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13(7):828–835
Ochoa AC, Zea AH, Hernandez C, Rodriguez PC (2007) Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res 13(2):721s–726s
Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK (1998) Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8(5):615–623
Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC (2005) Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med 202(7):931–939
Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB, Ochoa AC (2004) Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64(16):5839–5849
Sabel MS, Arora A, Su G, Chang AE (2006) Adoptive immunotherapy of breast cancer with lymph node cells primed by cryoablation of the primary tumor. Cryobiology 53(3):360–366
Sakai K, Yokote H, Murakami-Murofushi K, Tamura T, Saijo N, Nishio K (2007) Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci 98(9):1498–1503
Schluns KS, Lefrancois L (2003) Cytokine control of memory T-cell development and survival. Nat Rev Immunol 3(4):269–279
Serafini P, Borrello I, Bronte V (2006) Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol 16(1):53–65
Stoklasek TA, Schluns KS, Lefrancois L (2006) Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol 177(9):6072–6080
Tuttle TM, Inge TH, Bethke KP, McCrady CW, Pettit GR, Bear HD (1992) Activation and growth of murine tumor-specific T-cells which have in vivo activity with bryostatin 1. Cancer Res 52(3):548–553
Waldmann TA (2006) The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol 6(8):595–601
Weng NP, Liu K, Catalfamo M, Li Y, Henkart PA (2002) IL-15 is a growth factor and an activator of CD8 memory T cells. Ann N Y Acad Sci 975:46–56
Worschech A, Kmieciak M, Knutson KL, Bear HD, Szalay AA, Wang E, Marincola FM, Manjili MH (2008) Signatures associated with rejection or recurrence in HER-2/neu-positive mammary tumors. Cancer Res 68(7):2436–2446
Young MR, Lathers DM (1999) Myeloid progenitor cells mediate immune suppression in patients with head and neck cancers. Int J Immunopharmacol 21(4):241–252
Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC (2005) Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res 65(8):3044–3048
Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA (2007) IL-2 is essential for TGF-beta to convert naive CD4+ CD25- cells to CD25+ Foxp3+ regulatory T cells and for expansion of these cells. J Immunol 178(4):2018–2027
Zhou J, Zhong Y (2004) Breast cancer immunotherapy. Cell Mol Immunol 1(4):247–255
Acknowledgments
This work was supported by NIH R01 CA104757 grant (M. H. Manjili) and flow cytometry shared resources facility supported in part by the NIH grant P30CA16059. We thank Daniel Conrad and Jamie Sturgill for their assistance with purification of the anti-Gr1 antibody. We gratefully acknowledge the support of VCU Massey Cancer Centre and the Commonwealth Foundation for Cancer Research, Department of Defense Grant BC083048.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. K. Morales and M. Kmieciak have equally contributed to this work.
Rights and permissions
About this article
Cite this article
Morales, J.K., Kmieciak, M., Graham, L. et al. Adoptive transfer of HER2/neu-specific T cells expanded with alternating gamma chain cytokines mediate tumor regression when combined with the depletion of myeloid-derived suppressor cells. Cancer Immunol Immunother 58, 941–953 (2009). https://doi.org/10.1007/s00262-008-0609-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-008-0609-z