
Abstract
The immune attack against malignant tumors require the concerted action of CD8+ cytotoxic T lymphocytes (CTL) as well as CD4+ T helper cells. The contribution of T cell receptor (TCR) αβ+ CD4− CD8− double-negative (DN) T cells to anti-tumor immune responses is widely unknown. In previous studies, we have demonstrated that DN T cells with a broad TCR repertoire are present in humans in the peripheral blood and the lymph nodes of healthy individuals. Here, we characterize a human DN T cell clone (T4H2) recognizing an HLA-A2-restricted melanoma-associated antigenic gp100-peptide isolated from the peripheral blood of a melanoma patient. Antigen recognition by the T4H2 DN clone resulted in specific secretion of IFN-γ and TNF. Although lacking the CD8 molecule the gp100-specifc DN T cell clone was able to confer antigen-specific cytotoxicity against gp100-loaded target cells as well as HLA-A2+ gp100 expressing melanoma cells. The cytotoxic capacity was found to be perforin/granzymeB-dependent. Together, these data indicate that functionally active antigen-specific DN T cells recognizing MHC class I-restricted tumor-associated antigen (TAA) may contribute to anti-tumor immunity in vivo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The knowledge of human Ag-specific TCRαβ+ CD4− CD8− double-negative (DN) T cells and their role in the regulation of immune reponses is limited. Recently, we have shown that these cells exist in the peripheral blood as well as lymph nodes of healthy humans [1]. We found that human DN T cells reveal a long proliferative history and comprise of an antigen-experienced (CD45RA− CCR7+) as well as a naïve (CD45RA+ CCR7−) subpopulation [1]. Primary human DN T cells exhibit a polyclonal TCR repertoire with a broad distribution of TCRVβ families. However, it remains unclear whether human peripheral DN T cells recognize MHC-I or MHC-II restricted epitopes and mediate Ag-specific target cell recognition in the absence of the CD4 or CD8 coreceptor.
Following the established paradigm, CD4+ T cells recognize antigens presented by MHC class-II molecules, whereas CD8+ T cells recognize antigens in an MHC class-I context [2, 3]. However, alternative ways of antigen recognition, e.g. MHC class-I restricted CD4+ T cells have been demonstrated [4]. There is evidence from the murine system that antigen-recognition of DN T cells is connected to MHC class-I restricted epitopes: in MRL/MpJ-lpr/lpr mice, that have a mutant Fas gene and abnormal accumulation of DN T cells, several studies have demonstrated that TCRαβ+ DN T cells can be derived from CD8+ T cells and require MHC class-I or MHC class-I-like selection [5, 6]. These data are supported by several reports indicating that DN T cells are more closely related to CD8+ than to CD4+ T cells, e.g. due to demethylation of the CD8 gene [5, 7] and cytotoxic activity which is similar to conventional CD8+ T cells [8, 9]. Moreover, anti-leukemic cytotoxic activity of murine TCRαβ+ DN T cells has been demonstrated by Young et al. [10].
CD8 is encoded by two distinct genes, CD8α and CD8β, and is expressed on the surface of T cells primarily as a CD8αβ heterodimer [11]. Two different functions of the CD8 coreceptor on a T cell have been proposed. First, to stabilize the TCR/peptide/MHC complex: the Ig domain of CD8 binds to the conserved α3 domain of MHC class-I molecules on the cell surface [12, 13]. Second, to localize intracellularly the Src kinase lck to the TCR/CD3 complex [14, 15]. Earlier reports on the requirement of CD8 in antigen recognition have often been contradictory: while some TCRs are able to bind pMHC complexes despite the presence of anti-CD8 antibodies or mutations in the CD8-binding site [16], other TCRs require CD8 stabilization [17, 18]. In a recent study, we could substantiate the hypothesis that the contribution of the structural versus the signaling role of CD8 in T cells depends on the affinity of the TCR [16]. While the intracellular lck-binding domain of CD8 is critical for enhanced T cell activation regardless of the relative strength of the TCR, the extracellular domain of CD8 seems to be critical for TCRs with lower affinity but not those with higher affinity.
In this study, we have characterized a TAA-specific (gp100) MHC class-I restricted human DN T cell clone isolated from the peripheral blood of a melanoma patient. Functional data provide clear evidence that this clone exhibits similar characteristics as antigen-specific CD8+ cytotoxic T cells, indicating that human DN T cells may contribute to anti-tumor immunity in vivo.
Materials and methods
Media, cytokines and peptides
T cells were cultured in RPMI-1640 medium (Gibco, Karlsruhe, Germany) plus 10% human AB serum (PAN Biotech, Aidenbach, Germany). The following recombinant human cytokines were used: 800 U/ml granulocyte–macrophage colony-stimulating factor (GM-CSF; Schering-Plough, Brussels, Belgium), 500 U/ml IL-4, 5 ng/ml transforming growth factor-β1 (TGF-β1) (both from Tebu, Offenbach, Germany), 10 ng/ml IL-1β, 1,000 U/ml IL-6, 10 ng/ml tumor necrosis factor (TNF), 2,000 U/ml interferon-γ (IFN-γ), (all from PromoCell, Heidelberg, Germany), and 1 μg/ml prostaglandin E2 (PGE2; Pharmacia, Erlangen, Germany). Preparation of T cell growth factor (TCGF) was described previously [19]. The following HLA-A2-binding peptides were prepared by Clinalfa (Laeufelfingen, Switzerland): gp100209–217 (ITDQVPFSV) and Melan-A26–35 peptide (ELAGIGILTV). The identity of each peptide was confirmed by mass spectral analysis.
MHC peptide tetramers and mAbs
Phycoerythrin (PE)-conjugated HLA-A*0201 tetramer (TM) that had been folded around Melan-A26–35 or gp100209–217, were synthesized by Beckman Coulter (Fullerton, CA). For phenotypic analysis we used the following monoclonal antibodies (mAbs): α-CD8-APC, α-CD69-FITC, α-CD25-PE, α-CD45RO-PE, α-CD95-PE, α-HLA-DR-FITC, α-CD3-FITC, α-CD4-PerCP, α-TCRγδ-FITC (all from Becton Dickinson/Pharmingen, Heidelberg, Germany), α-CD28-FITC and α-CD62L-FITC (both from Caltag), α-CCR7-FITC (R&D Systems, Minneapolis), α-TCRαβ-PE, α-CD71-FITC (Immunotech/Beckman Coulter, Marseille, France). For intracellular staining the following unconjugated mAbs were used: α-CD8α-chain (Becton Dickinson), α-CD8β chain (Immunotech/Beckman Coulter), α-perforin and α-granzyme-B and (both from Hoelzel Diagnostika, Cologne, Germany). For blocking of the TCR, an unconjugated α-TCRVβ17 mAb (Immunotech) was used.
Cell lines
Melanoma cell lines Mel1300 (HLA-A2+, gp100+) and Na8 (HLA-A2+, gp100−) [19] were cultured in complete medium supplemented with 10% FCS (PAA, Pasching, Austria). The transporter associated with antigen processing (TAP)-deficient cell line T2 [21] was cultured in complete medium supplemented with 10% FCS. Prior to antigen-loading, T2 cells were kept for 6 h at room temperature (RT) in X-Vivo15 medium (Cambrex, Verviers, Belgium) and pulsed O/N exogenously with 5 μg/ml of the Melan-A or the gp100 peptide and 10 μg/ml β2-microglobulin (Scipac, Sittingbourne, UK) in serum-free complete medium. Ag-loaded T2 cells were washed four times with complete medium to remove redundant free peptide.
Generation and expansion of T cell lines and T cell clones
For the generation of CD8+ Melan-A/gp100-specific cytotoxic T cell (CTL) lines and clones, peripheral blood mononuclear cells (PBMC) from healthy donors were collected by leukapheresis, followed by density gradient centrifugation (Biocoll, Biochrom, Berlin). The study was approved by the University of Regensburg institutional review board. Informed consent was provided accordingly to the Declaration of Helsinki. Melan-A/gp100-specific CTL lines were generated as described previously [22]. Briefly, CD8+ T cells were enriched using a negative isolation strategy (Miltenyi Biotec, Bergisch-Gladbach, Germany). Autologous dendritic cells (DC) were generated from monocytes and cultured in complete medium plus 10% FCS, supplemented with GM-CSF, IL-4 and TGF-β. On day 6, fresh medium containing GM-CSF, IL-4, TNF, IL-6, IL-1β and PGE2 was added. The culture was continued for additional 48 h and mature DC were pulsed with 30 μg/ml of HLA-A2-binding Melan-A or gp100 peptide and 10 μg/ml human β2-microglobulin for 2 h at 37°C in serum-free complete medium. CD8+ T cells were restimulated weekly with antigen-pulsed DC in 96-well U-bottom plates. Complete medium supplemented with 3% TCGF was replenished twice a week. T cell clones were isolated from primary CTL lines by single cell sorting (FACS Aria, BD Biosciences, San Diego, CA) using the appropriate MHC-tetramers. CD8+ T cell clones were expanded by repetitive stimulation with allogeneic irradiated EBV-transformed B cells, irradiated allogeneic PBMC and PHA-L (1 mg/ml, Sigma, Steinheim, Germany).
The gp100-reactive T cell clone T4H2 was isolated from the peripheral blood of melanoma patient that had received the gp100:209–217 210 M peptide vaccine in incomplete Freund’s adjuvant as part of a clinical trial in the Surgery Branch, NCI. The T cell clone was maintained in X-Vivo 15 media (Biowhittaker, Walkersville, MD) supplemented with 10% human AB serum (Valley Biomedical, Winchester, VA), 2 mM l-glutamine (Mediatech, Herndon, VA), 100 IU/ml penicillin (Mediatech), 100 mg/ml streptomycin (Mediatech), and 300 IU rhIL-2 (Chiron Co., Emeryville, CA) per ml.
Intracellular cytokine staining, cytokine bead array
Prior to flow cytometric analysis, cells were stained with PE-coupled MHC-tetramers (30 min/37°C), then with mAbs and finally with 7AAD (BD/Pharmingen) for 15 min at 4°C in the dark. For intracellular mAb staining cells were fixed with 0.25% paraformaldehyde (PFA, Merck, Darmstadt, Germany), permeabilized with 10% saponine (Riedel-de-Haen, Seelze, Germany) in 5% human albumine (Octapharma, Langenfeld, Germany) and stained with the respective mAb. For quantification of secreted IL-2, IL-4, IL-5, IL-10, TNF and IFN-γ, DN and CD8+ T cells were stimulated at a T cell:stimulator cell ratio of 3:1 for different time points. The supernatant was collected and analyzed via the Th1/Th2 cytokine bead array kit (BD/Pharmingen) following the manufacturer’s instructions. Flow cytometry was performed on a FACSCalibur flow cytometer (BD/Biosciences). Data were analyzed with CellQuest (BD/Biosciences) software.
Complementary-determining region 3 size analysis of TCR Vβ transcripts
The complementary-determining region-3 (CDR3) of the polymerase chain reaction (PCR)-amplified TCR Vβ1-24 transcripts was analyzed using a run-off procedure [23]. The run-off products were run on an automated sequencer in the presence of fluorescent size markers. The length of DNA fragments and the fluorescence intensity of the bands were analyzed by GENESCAN672 software (Applied Biosystems, Foster City, USA).
51Cr-release assay
Conventional 51Cr-release assays were performed as previously described [20]. Briefly, the cytotoxicity of T cell lines was determined using triplicate cultures in V-bottomed plates. Target cells were labeled with 200 μCi for 1 h. Target cells were then washed twice and seeded into the plates at effector:target (E/T) ratios from 1:1 to 25:1 with 2,000 target cells/well. For spontaneous release, targets were plated without T cells in complete medium. For maximum release, target cells in 100 μl of medium were plated with 100 μl of 0.15% Triton-X-100 (Sigma). For blocking of perforin-mediated cytotoxicity, T cells were incubated O/N with Concanamycin-A (CMA, Sigma) at a final concentration of 115 nM, washed twice and added to the cytotoxicity assay. For blocking of granzyme-B the blocker z-AAD (Calbiochem/Merck, Darmstadt, Germany) was added at indicated concentrations to the 51Cr-release assays.
Reverse transcription: quantitative LightCycler PCR
Total RNA was isolated from cultured CD8+ T cells or DN T cells in 30 μl end volume using RNeasy Spin Columns from Quiagen (Hilden, Germany). Reverse transcription was performed using a SuperScriptTM II RNase H-Reverse Transcriptase (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s instructions; 0.25 μg total RNA from cultured CD8+ and DN T cells was reverse transcribed in a total volume of 20 μl. cDNA was amplified on a Roche Applied Science LightCycler using the QuantiTectTM SYBR Green PCR Kit (Qiagen, Hilden, Germany). The sequences of the primers used for amplification were as follows: sense strand 5′-ATG TGG CAG TGT CTG CTG AG-3′ and antisense strand 5′-GAT GCC TAG CCC AAT GAA AA-3′ for CD4; sense strand 5′-CAA TCT CAC AAG CGT GAA GCC-3′ and antisense strand 5′-GAA GGA AAT CAA CCA CAC TCA GC-3′ for CD8β-chain; sense strand 5′-CCC TGA GCA ACT CCA TCA TGT-3′ and antisense strand 5′-GTG GGC TTC GCT GGC A-3′ for CD8α-chain; sense strand 5′-ACC GAT TGG ATG GTT TAG TGA G-3′ and antisense strand 5′-CCT ACG GAA ACC TTG TTA CGA C-3′ for 18S RNA. Two microliters of cDNA was amplified in a total volume of 20 μl. Quantitative PCR consisted of 45 cycles, with denaturing at 95°C for 15 s, annealing at 57°C for 20 s, and extension at 72°C for 20 s.
DNA preparation, bisulfite conversion, PCR and sequencing
Genomic DNA was isolated using the DNeasy Blood & Tissue kit (Qiagen) following the protocol for cultured cells. Bisulfite conversion was performed using the EpiTect Bisulfite kit (Qiagen) according to the manufacturer’s instruction. After bisulfite modification, DNA was eluted in 20 μl buffer EB. PCR was performed using FastStart Taq polymerase (Roche) in a final volume of 50 μl containing 10 μl bisulfite-treated genomic DNA at 93°C for 5 s, and 40 cycles of 93°C for 15 s, 53°C for 15 s and 72°C for 70 s, and a final extension step of 5 min at 72°C. The sequences of CD8-specific primers were as follows: sense strand 5′-GTT TTT GGA GGA TGT GAT GTT ATT-3′ and antisense strand 5′-TTC AAC CCT CAA TAC CCA TTT TAC-3′. PCR products were cloned using TOPO2.1 (Invitrogen, Karlsruhe, Germany) and inserts of several individual clones were sequenced by GENEART (Regensburg, Germany).
Results
Phenotypic characterization of a human gp100-reactive DN T cell clone
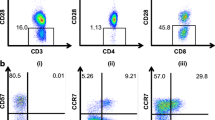
Here, we characterize the phenotype and function of a human TCRαβ+ DN T cell clone (T4H2) recognizing the HLA-A2 binding melanocyte differentiation antigen gp100209–217 (see Fig. 1a for gp-100 tetramer binding). As shown in Fig. 1a the surface phenotype of T4H2 cells is as follows: CD4−, CD8−, CD3+, TCRαβ+, CD69+, CD71dim, CD95+, CD25+, CD45RO+, CCR7−, CD62L−, and CD28−. With the exception of CD28, which was not expressed on T4H2 cells, DN T cells exhibit the same pattern of surface molecules as other conventional CD8+ gp100-specific CD8+ T cell clones (e.g. CD8+ clone 3.9, Fig. 1a). T4H2 cells were negative for CD27, CD62L, and expressed high levels of activation markers such as HLA-DR and CD95 (data not shown). In order to define the TCRVβ subfamily, we analyzed the complementary-determining region-3 (CDR3) size of T4H2 DN T cells using the spectratyping method. We found that the TCR belongs to the TCRVβ17 family (Fig. 1b). This finding could be verified by flow cytometry using an anti-TCRVβ17 mAb (see Fig. 1a). NK markers, such as CD16, NKG2D, CD94, CD158a (Fig. 1c) were completely absent with the exception of CD56 which was slightly expressed.

Phenotype of the gp100-reactive DN T cell clone T4H2. T4H2 cells were isolated from a melanoma patient after gp100-peptide vaccination and expanded over several months by repetitive stimulation with allogeneic feeder cells. A classical gp100-reactive CD8+ CTL clone (3.9), isolated from an HLA-A2+ healthy donor, served as a control. a T4H2 and 3.9 cells were harvested and stained with a panel of mAbs recognizing CD3, CD4, CD8, TCRαβ, TCRVβ17 (for T4H2) or TCRVβ7 (for 3.9), CD28, CD45RO, CCR7, CD69, CD25, CD71 and the HLA-A2/gp100-tetramer and analyzed by flow cytometry. Histograms are shown on gated lymphocytes (by forward and side scatter) to exclude residual feeder cells and fluorescence intensity is presented by the gray line. Open histograms show the respective isotype controls. b Analysis of the TCRVβ family distribution in T4H2 DN T cells via the TCR CDR3 spectratyping method. A clonal T cell population is represented by a distinct peak in the appropriate TCRVβ17 receptor family. One negative (TCRVβ16) and the TCRVβ17 positive run off results are shown. c Analysis of NK cell marker expression on T4H2 cells. T4H2 cells were stained with mAbs against NK cell markers CD56, CD158a, NKG2D, CD94, CD16, CD161, and analyzed by flow cytometry
We next compared the ability of DN T cells and CD8+ T cells to bind gp100 MHC-tetramers and found that the MHC-TM binding capacity is comparable in both clones (Fig. 1a). These data indicate that despite the lack of the CD8 molecule, the TCR binding and stabilization to the pMHC is not diminished in the DN T4H2 clone.
There is data suggesting that TCRαβ+ DN T cells may be closely related to CD8+ T cells [5, 7–9]. Thus, we aimed to elucidate whether the T4H2 clone belongs to the DN T-cell family or to CD8+ T cells not expressing the CD8 chain on their surface. First, we determined the intracellular CD8α- and CD8β-chain expression via flow cytometry. In contrast to CD8+ T cell lines (positive control), no significant CD8 protein expression could be detected in T4H2 cells (Fig. 2a). In line with these data, surface antibody staining revealed that no CD8α or -β protein is present on the surface of T4H2 cells (Fig. 1a).

Lack of CD8 coreceptor expression on the T4H2 DN T cell clone. a Intracellular expression of the CD8α chain and CD8β chain on the DN clone T4H2. For intracellular mAb staining cells were fixed with 0.25% paraformaldehyde, permeabilized with 10% saponine and stained with the respective anti-CD8 mAbs and analyzed by flow cytometry. A CD8+ Melan-A-specific CTL line served as a positive control. Histograms are shown on gated lymphocytes (by forward and side scatter) and fluorescence intensity is presented by the gray line. Open histograms show the isotype controls. b Quantitative light cycler PCR was performed to detect CD8α-chain, CD8β-chain and CD4 mRNA in T4H2 cells. Values were normalized to the amount of CD8 transcripts either in Melan-A specific CD8+ CTL lines (CTL#492) or CD4 transcripts in freshly isolated CD4+ T cells. Mean values ± SEM from triplicates are shown
In order to exclude that T4H2 DN T cells express low amounts of CD8 molecules we next determined CD8α- or CD8β-chain mRNA transcripts using quantitative Real Time PCR. As shown in Fig. 2b, we could not detect any CD8 mRNA in the DN T cell clone in contrast to CD8+ CTL cell lines or the CD8+ T cell clone 3.9. As expected, there is also no CD4 mRNA detectable in the T4H2 DN T cell clone compared to isolated CD4+ T cells (Fig. 2b).
To investigate whether DNA methylation might play a role in the silencing of CD8, we analyzed the methylation status of the CD8α promoter region in DN T cells as well as CD8+ and CD4+ T cells (Fig. 3). Although the degree of methylation observed in the DN T cell clone was higher than in CD8+ or CD4+ T cells, the majority of CpG residues remained free of methylation (appr. 12% mCpG compared to 1% mCpG in CD8+ T cells). Therefore, it is unlikely that CpG methylation of the proximal promoter plays a major role in CD8 silencing of the DN T cells.

CD8 gene demethylation in T4H2 DN T cells. Schematic overview of human CD8α promoter and positions of CpG motifs (upper panel). Genomic DNA from DN T cell clone T4H2, CD8+ T cell clone 3.9 and a CD4+ T cell clone was analyzed by bisulfite sequencing. In the matrix, each row represents one sequenced insert, each box shows methylation status of a single CpG. Filled boxes indicate methylation (lower panel)
Functional characterization of the T4H2 DN T cell clone
Moreover, we tested the specific cytokine pattern of T4H2 DN T cells upon MHC class-I restricted Ag-specific recognition. Therefore, we determined the secretion of IL-2, IL-4, IL-5, IL-10, TNF, and IFN-γ in the supernatant of T4H2 cells after stimulation with either gp100- or control peptide (Melan-A)-pulsed T2 target cells, or with the gp100-expressing melanoma cell line Mel1300. The gp100-specific CD8+ T cell clone 3.9 served as a control (see Fig. 4 for 4 h supernatants, 24 h data not shown). We found that the T4H2 DN T cell clone predominantly secretes IFN-γ and TNF upon gp100-specific recognition whereas unstimulated T4H2 cells (data not shown) or T4H2 cells stimulated with Melan-A-pulsed T2 cells did not secrete any cytokines. While the total amount of cytokines, secreted by the specifically stimulated DN T cell clone T4H2 tends to be lower compared to conventional CD8+ T cells, the ratio between IFN-γ and TNF was similar to that of the CD8+ T cell clone. Please note, that the melanoma cell line Mel1300 produced low amounts of IL-10.

gp100-reactive DN T cells secrete cytokines upon recognition of the target cells. Cytokine profile of the T4H2 DN clone and the CD8+ T cell clone 3.9 after a 4 h coculture with either gp100- or Melan-A (MelA)-pulsed T2 cells or HLA-A2+, gp100+ expressing melanoma cells (Mel1300). Melan-A loaded T2 cells were used as control stimulator cells. The amount of cytokines in the supernatant was determined by the Cytometric Bead Array (CBA). Data show mean ± SEM of three independent experiments
We next asked whether the T4H2 DN T cell clone exerts any cytotoxic activity against gp100 expressing target cells although lacking CD8. We found that the T4H2 DN T cell clone exerts an antigen-specific cytotoxic activity against gp100-loaded target cells while T2 cells loaded with an irrelevant peptide (Melan-A) were not affected (Fig. 5). In order to clarify, if T4H2 DN T cells also kill tumor cells expressing endogenously processed gp100 epitopes, we used different gp100+ and gp100−, HLA-A2+ melanoma cell lines as target cells. We found potent cytotoxic activity against the gp100+ tumor cell line Mel 1300 (Fig. 5, filled triangles), while the gp100− cell line Na8 was not recognized at all (filled circles). Blocking of the TCR on DN T cells with an α-TCRVβ17 mAb completely abrogated target cell killing (Fig. 5, open triangles). Together, these data clearly demonstrate that the T4H2 DN T cell clone mediates a TCR-driven Ag-specific target cell killing. Next, we performed 51Cr-release assays with the T4H2 DN T cell clone and the gp100-specific CD8+ T cell clone 3.9 as a control against T2 cells loaded with different concentrations of the gp100 peptide. We found that the T4H2 DN T cell clone exhibits a significant cytotoxicity against T2 cells loaded with a peptide concentration of 5 μg/ml, albeit the absolute amount of cell lysis was lower compared to CD8+ T cells (Fig. 6). Dilution of the gp100 peptide to 0.5 μg/ml led to a significant reduction of the DN T cell-mediated cytotoxicity, while the CD8+ T cell clone was still able to mediate the full cytotoxicity up to a concentration of 0.05 μg/ml.

Cytotoxic activity of T4H2 DN T cells against endogenously presented gp100 antigen on melanoma cells. The cytotoxic capacity of the T4H2 DN T cell clone was assessed by a standard 4 h 51Cr-release assay. T2 cells loaded exogenously with gp100 peptide (filled squares) as well as the gp100-expressing melanoma cell line Mel1300 (filled triangles) served as target cells. As negative controls Melan-A-loaded T2 (open squares) and the melanoma cell line Na8 (HLA-A2+, gp100−, filled circles) were used. For TCR blocking studies, effector cells were preincubated for 30 min with a neutralizing anti-TCRVβ17 mAb (open triangles) or the isotype control (open circles), washed and coincubated with Mel1300 cells. Data represent means ± SEM of triplicates

Cytotoxic activity of the gp100-reactive DN (T4H2) and CD8+ (3.9) T cell clone was examined at an effector-to-target (E/T) ratio of 5:1 using indicated concentrations of gp100 peptide loaded onto T2 cells. The cytotoxic capacity of the T cell clones was assessed by a standard 4 h 51Cr-release assay. Shown are mean values of triplicates ±SEM
In order to elucidate the mechanism of target cell killing in the DN T4H2 T cell clone, we analyzed the presence of the effector molecules perforin and granzyme-B by intracellular mAb staining. We found high expression of perforin and granzyme-B in T4H2 comparable to CD8+ CTL lines and CD8+ T cell clones (Fig. 7a). Moreover, we studied the functional impact of both effector molecules using Concanamycin-A (CMA), an inhibitor of perforin and z-AAD, an inhibitor of granzyme-B. Pretreatment of T4H2 cells with CMA was able to completely abolish the cytotoxic activity of T4H2 cells similar to that observed in CD8+ CTL (see Fig. 7b). In line with these data, z-AAD revealed a dose-dependent inhibition of target cell killing by T4H2 cells (Fig. 7b). Thus, both effector molecules are involved in antigen-specific target cell killing mediated by the T4H2 DN T cell clone.

Death pathway of T4H2 clone mediated cytotoxicity. a T4H2 cells were harvested, stained intracellularly with mAbs recognizing human effector molecules perforin and granzyme-B (gzm-B) (gray histograms) or with an isotypic control (open histograms), and analyzed by flow cytometry. The gp100-specific CD8+ T cell clone 3.9 as well as a Melan-A specific CD8+ CTL line (CTL line) served as a positive control. b Inhibition of the T4H2 cell-mediated target cell killing by concanamycin-A (CMA, inhibitor of perforin-mediated killing) and z-AAD (granzyme-B inhibitor). T4H2 cells were cultured with CMA or z-AAD. Cytotoxic activity was examined at an E/T ratio of 5:1 using the gp100 expressing melanoma cell line Mel1300. Data are shown as means of triplicates in percentage of unblocked control ±SEM
Discussion
Here, we have characterized a gp100-specific TCRαβ+ CD4− CD8− DN T cell clone (T4H2) isolated from the peripheral blood of a melanoma patient. T4H2 cells exhibit a very stable phenotype, lacking CD4 and CD8 expression on the protein and mRNA level. The phenotypic surface marker pattern (CD3+, CD69+, CD95+, CD25+, CD45RO+, CCR7−, CD62L−, CD27−, CD28−) indicates that these cells are highly activated effector-memory T cells being identical to conventional CD8+ T cell clones with the exception of CD28 which was negative on DN T cells. Yet, it is unknown whether T4H2 cells are susceptible to other costimulatory signals as it has been reported for conventional T lymphocytes [e.g. CD27, ICOS, OX40, 4-1BB [24–27]]. However, the lack of costimulation via CD28 rises the formal question whether the T4H2 cells having an activated phenotype upon ex vivo expansion would have been activated similarly under in vivo conditions. Nevertheless, it is of interest that this DN T cell clone is able to exert cytotoxic effector functions despite the lack of CD8 and CD28. This phenomenon may be explained by a high affinity of its TCR against the antigen.
T4H2 cells exhibit similar biological functions compared to conventional CD8+ CTL: first, the cytokine pattern of T4H2 cells was dominated by IFN-γ and TNF secretion, which was similar to the gp100-reactive CD8+ T cells used as a control. These results are also in line with previous findings from freshly isolated DN T cells predominantly secreting IFN-γ [1]. Second, despite the lack of the CD8 coreceptor, T4H2 DN T cells exhibit a strong TCR-dependent Ag-specific cytotoxic activity. This finding is in line with our former observations of CD4+ T cells mediating efficient reactivity against MHC class-I restricted antigens without a significant contribution of coreceptors [4]. Additional studies with a Hepatitis C virus-reactive TCR cloned into Jurkat cells came to a similar conclusion [28]. Furthermore, it has been reported that retroviral transfer of the TCRαβ into TCRγδ+ T cells is sufficient to convey high cytotoxic activity against leukemic cells [29]. The perforin/granzyme-B based death pathway of the T4H2 clone is contradictory to murine TCRαβ+ DN T cells, which mediate cytotoxicity mainly via the fas/fasL pathway [10, 30]. However, recent publications report that murine xenoreactive TCRαβ+ DN T cells are also able to kill target cells via the perforin/granzyme-B pathway [31, 32].
Previous findings from our own and other groups corroborate the hypothesis that any TCR that is capable to bind peptide/MHC complexes independently of CD8 is of a higher relative affinity than TCRs that require CD8 for binding [33–35]. Our latest studies indicating that the CD8 coreceptor is mainly necessary to support low affinity TCRs [36] further suggests that the T4H2 TCR is functionally active due to its high affinity towards the gp100 antigen. The proof that high affinity TCRs are able to confer specificity and functional activity has also been given by Kuball et al. [37]. They could convincingly show that the gene transfer of a high affinity TCR recognizing the tumor suppressor protein p53 can redirect CD4+ T lymphocytes.
One open question remaining is the selection of this HLA-A2 restricted DN T cell clone in the thymus. Current models of thymic selection require that MHC class-I peptide molecules encoded by the thymus positively select classical CD8+ T cells [38]. However, there are examples where TCR αβ DN T cells proceed through thymic selection leading to functional T cells in the periphery. Van Laethem et al. [39] reported functional TCR αβ DN T cells in CD4/CD8/β2 m/I-A quad deficient mice. Recently, we generated a transgenic mouse using the TIL1383I TCR which recognizes the HLA-A2 restricted tyrosinase368–376 peptide (unpublished). In HLA-A2 transgenic mice, the TIL 1383I TCR is expressed in CD4 single positive (SP), CD8 SP, and DN T cells in the periphery. These T cells function since the mice get spontaneous vitiligo starting at 4–5 weeks of age (unpublished). While the selection process in the thymus leading to mature peripheral DN T cells is not yet understood, these T cells clearly exist and there are animal models now available to study this process in detail.
In this study, we have shown that the CD8α promoter in the DN T cell clone is poorly methylated. Similar data were found in DN T cells from mice lacking expression of fas (lpr) or fas-ligand (gld) [40, 41]. More recently, Bristeau-Leprince et al. [42] showed that DN T cells from ALPS patients share with CD8+ T cells unique CDR3 sequences across several TCRVβ families, arguing for the CD8 origin of DN T cells. However, Ford et al. [43] demonstrated that DN T cells do not mature from CD8+ T cell precursors, nor CD8 expression is required for their development in vivo. Taken together, our data suggest that CD8 is not aberrantly silenced by DNA methylation and that the DN T cell clone likely progressed through a CD8-expressing stage. However, we cannot exclude the possibility that the clone may have lost CD8 expression during its in vitro isolation from the PBMC population. Unfortunately, a clear cut identification of the gp100-specific DN clonotype in the small DN peripheral blood lymphocyte population was impossible because of the lack of material from the patient, who passed away.
Another possible mechanism of thymic maturation is that a high relative TCR affinity compensates for the lack of CD8 expression. Moreover, negative selection of conventional T cells is strongly supposed to be promoted by B7-1/B7-2-mediated signals [44, 45]. As mentioned above, we found that the T4H2 clone lacks a CD28 surface expression. Thus, it seems likely that the omitted negative selection of the T4H2 clone was at least partially caused by an absent B7-1/B7-2-mediated costimulation. Interestingly, we could show in former studies that a lack of CD28 expression is characteristic for the majority of human TCRαβ+ DN T cells found in the periphery [1].
Our findings support previous results with transduced cells from our own [28] and other groups [29] indicating that CD8-independent MHC class-I driven cytotoxicity is possible. In line with earlier reports of MHC class-I restricted cytotoxic CD4+ T lymphocytes [4] we conclude that TCRαβ+ DN T cells are able to contribute to anti-tumor immune responses in vivo. Only limited data are available regarding the homing and tissue distribution of antigen-specific DN T cells. Thus, it remains unclear whether TCRαβ+ DN T cells are present at the tumor site and how they are activated. Moreover, it is still open whether these cytotoxic TCRαβ+ DN T cells are related to previously described regulatory TCRαβ+ DN T cells [1, 30].
Further experiments are warranted to determine the impact of non-classical T cell populations for anti-tumor immunity.
References
Fischer K, Voelkl S, Heymann J, Przybylski GK, Mondal K, Laumer M, Kunz-Schughart L, Schmidt CA, Andreesen R, Mackensen A (2005) Isolation and characterization of human antigen-specific TCR alpha beta+ CD4–CD8- double-negative regulatory T cells. Blood 105:2828–2835
Zinkernagel RM, Doherty PC (1997) The discovery of MHC restriction. Immunol Today 18:14–17
Sprent J, Schaefer M, Lo D, Korngold R (1986) Functions of purified L3T4+ and Lyt-2+ cells in vitro and in vivo. Immunol Rev 91:195–218
Nishimura MI, Avichezer D, Custer MC, Lee CS, Chen C, Parkhurst MR, Diamond RA, Robbins PF, Schwartzentruber DJ, Rosenberg SA (1999) MHC class I-restricted recognition of a melanoma antigen by a human CD4+ tumor infiltrating lymphocyte. Cancer Res 59:6230–6238
Takahama Y, Kosugi A, Singer A (1991) Phenotype, ontogeny, and repertoire of CD4–CD8- T cell receptor alpha beta+ thymocytes. Variable influence of self-antigens on T cell receptor V beta usage. J Immunol 146:1134–1141
Herron LR, Eisenberg RA, Roper E, Kakkanaiah VN, Cohen PL, Kotzin BL (1993) Selection of the T cell receptor repertoire in Lpr mice. J Immunol 151:3450–3459
Landolfi MM, Van Houten N, Russell JQ, Scollay R, Parnes JR, Budd RC (1993) CD2–CD4–CD8- lymph node T lymphocytes in MRL lpr/lpr mice are derived from a CD2+ CD4+ CD8+ thymic precursor. J Immunol 151:1086–1096
Hammond DM, Nagarkatti PS, Gote LR, Seth A, Hassuneh MR, Nagarkatti M (1993) Double-negative T cells from MRL-lpr/lpr mice mediate cytolytic activity when triggered through adhesion molecules and constitutively express perforin gene. J Exp Med 178:2225–2230
Cai Z, Sprent J (1994) Resting and activated T cells display different requirements for CD8 molecules. J Exp Med 179:2005–2015
Young KJ, Kay LS, Phillips MJ, Zhang L (2003) Antitumor activity mediated by double-negative T cells. Cancer Res 63:8014–8021
Ledbetter JA, Seaman WE, Tsu TT, Herzenberg LA (1981) Lyt-2 and lyt-3 antigens are on two different polypeptide subunits linked by disulfide bonds. Relationship of subunits to T cell cytolytic activity. J Exp Med 153:1503–1516
Newberg MH, Ridge JP, Vining DR, Salter RD, Engelhard VH (1992) Species specificity in the interaction of CD8 with the alpha 3 domain of MHC class I molecules. J Immunol 149:136–142
Wedemeyer H, He XS, Nascimbeni M, Davis AR, Greenberg HB, Hoofnagle JH, Liang TJ, Alter H, Rehermann B (2002) Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol 169:3447–3458
Salter RD, Norment AM, Chen BP, Clayberger C, Krensky AM, Littman DR, Parham P (1989) Polymorphism in the alpha 3 domain of HLA-A molecules affects binding to CD8. Nature 338:345–347
Rudd CE, Anderson P, Morimoto C, Streuli M, Schlossman SF (1989) Molecular interactions T-cell subsets and a role of the CD4/CD8:p56lck complex in human T-cell activation. Immunol Rev 111:225–266
Muller D, Pederson K, Murray R, Frelinger JA (1991) A single amino acid substitution in an MHC class I molecule allows heteroclitic recognition by lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes. J Immunol 147:1392–1397
Garcia KC, Scott CA, Brunmark A, Carbone FR, Peterson PA, Wilson IA, Teyton L (1996) CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature 384:577–581
Daniels MA, Jameson SC (2000) Critical role for CD8 in T cell receptor binding and activation by peptide/major histocompatibility complex multimers. J Exp Med 191:335–346
Mackensen A, Carcelain G, Viel S, Raynal MC, Michalaki H, Triebel F, Bosq J, Hercend T (1994) Direct evidence to support the immunosurveillance concept in a human regressive melanoma. J Clin Invest 93:1397–1402
Meidenbauer N, Zippelius A, Pittet MJ, Laumer M, Vogl S, Heymann J, Rehli M, Seliger B, Schwarz S, Le Gal FA, Dietrich PY, Andreesen R, Romero P, Mackensen A (2004) High frequency of functionally active Melan-a-specific T cells in a patient with progressive immunoproteasome-deficient melanoma. Cancer Res 64:6319–6326
Salter RD, Howell DN, Cresswell P (1985) Genes regulating HLA class I antigen expression in T–B lymphoblast hybrids. Immunogenetics 21:235–246
Meidenbauer N, Marienhagen J, Laumer M, Vogl S, Heymann J, Andreesen R, Mackensen A (2003) Survival and tumor localization of adoptively transferred Melan-A-specific T cells in melanoma patients. J Immunol 170:2161–2169
Pannetier C, Even J, Kourilsky P (1995) T-cell repertoire diversity and clonal expansions in normal and clinical samples. Immunol Today 16:176–181
Hendriks J, Xiao Y, Borst J (2003) CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med 198:1369–1380
van Berkel ME, Oosterwegel MA (2006) CD28 and ICOS: similar or separate costimulators of T cells? Immunol Lett 105:115–122
Weinberg AD, Vella AT, Croft M (1998) OX-40: life beyond the effector T cell stage. Semin Immunol 10:471–480
Cheuk AT, Mufti GJ, Guinn BA (2004) Role of 4-1BB:4-1BB ligand in cancer immunotherapy. Cancer Gene Ther 11:215–226
Callender GG, Rosen HR, Roszkowski JJ, Lyons GE, Li M, Moore T, Brasic N, McKee MD, Nishimura MI (2006) Identification of a hepatitis C virus-reactive T cell receptor that does not require CD8 for target cell recognition. Hepatology 43:973–981
van der Veken LT, Hagedoorn RS, van Loenen MM, Willemze R, Falkenburg JH, Heemskerk MH (2006) Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res 66:3331–3337
Zhang ZX, Yang L, Young KJ, DuTemple B, Zhang L (2000) Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat Med 6:782–789
Zhang ZX, Ma Y, Wang H, Arp J, Jiang J, Huang X, He KM, Garcia B, Madrenas J, Zhong R (2006) Double-negative T cells, activated by xenoantigen, lyse autologous B and T cells using a perforin/granzyme-dependent, Fas–Fas ligand-independent pathway. J Immunol 177:6920–6929
Ma Y, He KM, Garcia B, Min W, Jevnikar A, Zhang ZX (2008) Adoptive transfer of double negative T regulatory cells induces B-cell death in vivo and alters rejection pattern of rat-to-mouse heart transplantation. Xenotransplantation 15:56–63
Roszkowski JJ, Lyons GE, Kast WM, Yee C, Van Besien K, Nishimura MI (2005) Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res 65:1570–1576
Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI (1999) Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol 163:507–513
Cole DJ, Weil DP, Shilyansky J, Custer M, Kawakami Y, Rosenberg SA, Nishimura MI (1995) Characterization of the functional specificity of a cloned T-cell receptor heterodimer recognizing the MART-1 melanoma antigen. Cancer Res 55:748–752
Lyons GE, Moore T, Brasic N, Li M, Roszkowski JJ, Nishimura MI (2006) Influence of human CD8 on antigen recognition by T-cell receptor-transduced cells. Cancer Res 66:11455–11461
Kuball J, Schmitz FW, Voss RH, Ferreira EA, Engel R, Guillaume P, Strand S, Romero P, Huber C, Sherman LA, Theobald M (2005) Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity 22:117–129
Urdahl KB, Sun JC, Bevan MJ (2002) Positive selection of MHC class Ib-restricted CD8(+) T cells on hematopoietic cells. Nature Immunol 3:772–779
Van Laethem F, Sarafova SD, Park JH, Tai X, Pobezinsky L, Guinter TI, Adoro S, Adams A, Sharrow SO, Feigenbaum L, Singer A (2007) Deletion of CD4 and CD8 coreceptors permits generation of alphabetaT cells that recognize antigens independently of the MHC. Immunity 27:735–750
Landolfi MM, van Houten N, Russell JQ, Scollay R, Parnes JR, Budd RC (1993) CD2–CD4–CD8- lymph node T lymphocytes in MRL lpr/lpr mice are derived from a CD2+ CD4+ CD8+ thymic precursor. J Immunol 151:186–1096
Wadsworth S, Yui K, Siegel RM, Tenenholz DE, Hirsch JA, Greene MI (1990) Origin and selection of peripheral CD4–CD8- T cells bearing alpha/beta T cell antigen receptors in autoimmune gld mice. Eur J Immunol 20:723–730
Bristeau-Leprince A, Mateo V, Lim A, Maqerus-Chatinet A, Solary E, Fischer A, Rieuy-Laucat F, Gouqeon ML (2008) Human TCR alpha/beta+ CD4–CD8- double-negative T cells in patients with autoimmune lymphoproliferative syndrome express restricted Vbeta TCR diversity and are clonally related to CD8+ T cells. J Immunol 181:440–842
Ford MS, Zhang ZX, Chen W, Zhang L (2006) Double-negative T regulatory cells can develop outside the thymus and do not mature from CD8+ T cell precursors. J Immunol 177:2803–2809
Buhlmann JE, Elkin SK, Sharpe AH (2003) A role for the B7-1/B7-2:CD28/CTLA-4 pathway during negative selection. J Immunol 170:5421–5428
Gao JX, Zhang H, Bai XF, Wen J, Zheng X, Liu J, Zheng P, Liu Y (2002) Perinatal blockade of b7-1 and b7-2 inhibits clonal deletion of highly pathogenic autoreactive T cells. J Exp Med 195:959–971
Acknowledgments
We are grateful to the patient who took part in this study. We thank Jana Berger for excellent technical assistance. The authors have no conflicting financial interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
A. Mackensen and K. Fischer contributed equally to this work and should be considered joint senior authors. This work was supported by the Deutsche Forschungsgemeinschaft (MA 1351/5-1, KFO 146) and NIH grants CA90873, CA102280, 104947 (MIN).
Companion paper: “Relationship between CD8-dependent antigen recognition, T cell functional avidity, and tumor cell recognition” by Tamson V. Moore et al. doi: 10.1007/s00262-008-0594-2.
Rights and permissions
About this article
Cite this article
Voelkl, S., Moore, T.V., Rehli, M. et al. Characterization of MHC class-I restricted TCRαβ+ CD4− CD8− double negative T cells recognizing the gp100 antigen from a melanoma patient after gp100 vaccination. Cancer Immunol Immunother 58, 709–718 (2009). https://doi.org/10.1007/s00262-008-0593-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-008-0593-3