
Abstract
Although many multiple myeloma (MM) patients initially respond to cytotoxic therapy, most eventually relapse. Novel therapeutic strategies employing a combination of chemotherapy with targeted biologics may significantly enhance the response of tumor cells to treatment. We tested a fully human anti-IGF-IR antibody (A12) against MM, and showed specific inhibition of IGF-I or serum -induced IGF-IR signaling in MM cells in vitro. The A12 as a single agent was demonstrated to exert modest to significant inhibition of tumor growth in vivo in various subcutaneous xenograft MM models. The A12 was also evaluated in a disseminated xenograft MM.1S NOD/SCID model as monotherapy or in combination with other drugs (bortezomib, melphalan) currently in clinical use. The tumor burden, as determined by luciferase bioimaging, was sharply decreased, and overall survival significantly prolonged when the therapies were combined. Immunohistochemical analysis demonstrated that the A12 treated tumors had significantly decreased vascularization compared to control tumors. Furthermore, most MM lines constitutively secreted significant quantities of VEGF, and this was enhanced following IGF-I treatment. Inhibition of IGF-IR by the A12 in vitro suppressed both constitutive and IGF-I-induced secretion of VEGF, indicating that a putative anti-angiogenic mechanism associated with the A12 treatment may contribute to its anti-tumor effect.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM) is a progressive B-lineage neoplasia characterized by the accumulation of malignant plasma cells in the bone marrow associated with extensive osteolytic bone destruction, renal failure, anemia, and hypercalcemia [1–3]. In spite of extensive research and the development of novel therapies, the MM remains an incurable malignancy. In addition to standard chemotherapy, high-dose chemotherapy with autologous stem cell transplantation improved survival [4]. However, all patients eventually relapse. In the past years, major advances have been made with the use of thalidomide and the arrival of promising novel approaches such as bortezomib against MM, however, 65% of patients with relapsed refractory disease do not respond to these agents [5].
The insulin-like growth factor I (IGF-I) has been implicated in carcinogenesis, promoting growth, migration, and survival of cancer cells in a wide range of human tumors [6]. Expression of the IGF-I receptor (IGF-IR) has been reported in a broad panel of tumor types, including the MM, colon, prostate, breast, ovarian, and pancreatic cancer [6–11]. The physiological ligands IGF-I/IGF-II are potent mitogens for diverse cancer cell lines in vitro [12–15]. High levels of IGF-I and IGF-II expression have been noted in tumors and associated stromal cells and may stimulate cancer cell growth in an autocrine or paracrine manner [6, 16]. Epidemiological studies have correlated high plasma levels of IGF-I with increased risk for prostate, colon, lung, and breast cancer [17–20]. In addition to its role in proliferation of cancer cells, signaling through IGF-IR protects cells from apoptosis caused by growth factor deprivation, anchorage independence, or cytotoxic drug treatment [21, 22].
The IGF-IR signaling has been extensively studied in MM cells [23–31]. Freund et al. [23] reported that the MM cell lines expressed more insulin receptors (IRs) and IGF-IRs than did Burkitt lymphoma and B lymphoblastoid lines. In concert with the MM growth factor IL-6, the IGF-I stimulated MM cell proliferation, prolonged cell survival, and protected against apoptosis [24–26]. Tucci et al. observed a correlation between levels of serum IGF-I secreted from a co-existing pituitary macroadenoma in a patient with MGUS, to progression of the monoclonal gammopathy to overt MM [27]. The IGF-IR signal transduction initiates from the binding of high-affinity ligands (IGF-I/ IGF-II) to extracellular α-subunits of IGF-IR, which results in trans-autophosphorylation of specific tyrosine residues in the β-subunits of IGF-IR. Activated IGF-IR phosphorylates its substrates which in turn activate downstream signaling pathways, including the mitogen activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3-K) pathways [22, 28, 29]. In addition, the IGF-I is also implicated in chemotaxis of human MM cells mediated via activation of PI3-K/AKT and β-integrin [30] or protein kinase D/protein kinase C micro (PKD/PKCmicro) and RhoA associated kinase [31].
The fully human monoclonal antibody A12 is a selective antagonist of the IGF-IR, that acts as an antagonist of ligand binding and signaling and is further capable of facilitating the degradation of the IGF-IR after uptake and internalization, leading to a reduction in surface receptor density on treated tumor cells [32]. The goal of the present study was to test the anti-tumor activity of this potent antagonistic anti-IGF-IR antibody on MM in vitro and in vivo. We demonstrated that antibody A12 is a potent inhibitor of exogenous IGF-I-induced, serum-stimulated, or autocrine mediated proliferation and survival in MM cell lines in vitro. Treatment with A12, either alone or in combination with other antitumor agents, inhibited the growth of subcutaneous and disseminated human MM xenografts in NOD/SCID mice and effected enhanced survival when administered in combination with conventional cytotoxic drug (Melphalan) or with proteasome inhibitor (bortezomib). The antibody exhibited strong anti-tumor activity in vitro and in vivo, indicating its potential as a candidate for clinical therapy of MM.
Materials and methods
Reagents and cell lines
Human recombinant insulin-like growth factor I (IGF-I) and monoclonal anti-human IGF-IR-fluorescein were purchased from R&D Systems Inc. A fully human monoclonal antibody (A12) to IGF-IR was obtained from ImClone Systems Incorporated [32]. Melphalan and Bortezomib were obtained from Glaxo Smith Kline and Millennium Pharmaceuticals Incorporated, respectively. All other chemicals were purchased from Sigma (St. Louis, MO, USA) unless otherwise specified. DNA modifying or restriction enzymes were from NEB (Beverly, MA, USA). Fetal calf serum (FCS) was obtained from HyClone (Logan, UT, USA). The myeloma MM.1S line was a generous gift from Dr. Nancy L. Krett (Robert H. Lurie Comprehensive Cancer Center, Northwestern University). The CAG cell line was kindly provided by Dr. J. Epstein (Myeloma Institute for Research and Therapy, University of Arkansas). The human bone marrow stroma-dependent cell line MOLP-5 was obtained from Dr. Yoshinobu Matsuo (Hayashibara Biochemical Labs, Inc., Japan). All other myeloma cell lines used in this study were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in RPMI1640 medium (InVitrogen) supplemented with 10% FCS and 2 mM glutamine, or QBSF medium (Quality Biological, Inc.) and maintained in humidified incubators at 37°C in 5% CO2.
Fluorescence-activated cell sorting (FACS)
The FACS analysis was performed on cells cultured for 24 h in RPMI1640 medium supplement with 2% FCS and 2 mM glutamine. Cells were washed twice with cold PBS, and resuspended in 2% bovine serum albumin/PBS, and incubated with monoclonal anti-IGF-IR fluorescein following manufacturer’s instructions. Monoclonal anti-mouse IgG1 fluorescein was used as an isotype control (R&D Systems). Nonspecific binding to Fc receptors was blocked by treatment with anti-Fc antibodies for 15 min at 4°C. Cells were incubated with antibodies at 4°C for 45 min. All FACS analyses were performed on a FACSCalibur (Becton Dickinson) and data were analyzed using FlowJo (Tree Star, Inc.). Results are expressed as the mean fluorescence intensity ratio of positive versus isotype control. Cells expressing eGFP were sorted on a MoFlo (DakoCytomation).
Western blotting and immunoprecipitation
Receptor phosphorylation experiments were performed as described previously [32]. Briefly, RPMI 8226 cells were serum-deprived for 24 h, followed by 2 h incubation with A12 or anti-KLH control antibody (ImClone Systems, New York, NY, USA). Cells were pulsed with IGF-I for 10 min and lysates prepared. Total IGF-IR was immunoprecipitated using the alpha subunit antibody 7B1 (ImClone Systems, New York, NY, USA). Western blots of the resulting immunoprecipitate were first probed with an anti-phospho-tyrosine antibody, stripped and reprobed with a C-20 (Santa Cruz Biotech, Santa Cruz, CA, USA) monoclonal against IGF-IR-β subunit. Western blot analysis of Akt and MAPK (ERK 1, 2) signaling was performed as described previously [32] using antibodies directed against phospho Akt (Upstate Cell Signaling Solutions, Charlottesville, VA, USA), total Akt (Cell Signaling Technology, Beverly, MA, USA), phospho-ERK 1, 2 (Upstate Cell Signaling Solutions), and total ERK 1, 2 (Cell Signalling Technology). For analysis of antibody-mediated receptor degradation, cells were washed and then fed with serum-depleted medium and incubated with either A12 or IGF-I for the times indicated. Radioimmunoprecipitation (RIPA) lysates were electrophoresed on 4–12% Bis-Tris polyacrylamide gels, transferred to a nitrocellulose membrane and IGF-IR detected using antibody C-20. Quantitation of relative band intensity was performed using an Alpha Imager system and supporting software (Alpha Innotech, San Leandro, CA, USA).
XTT assay
Cell proliferation and viability measurements were assessed by a colorimetric assay using sodium 39-(phenylaminocarbonyl)-3,4-tetrazolium-bis (4-methoxy-6-nitro) benzene sulfonic acid hydrate (XTT), and phenazine methosulphate (PMS). After serum starvation for 16 h, cells from indicated lines were plated at 5∼10 × 103/well in 96 well plates in the presence of different concentration of compounds for 52 h. Then 50 μL of XTT/PMS mixture was added to each well and incubated for an additional 20 h at 37°C. Plate absorbance was measured directly at optical density of 490 nm (reference wavelength at 650 nm) in an ELISA reader (Thermo Max Microplate Reader, Molecular Devices, CA, USA). The data were expressed as the mean of readings obtained in treated wells with subtraction of the control reading.
Lentiviral vectors construction, production, and transduction
To construct a lentiviral vector to express a fusion protein of enhanced green fluorescent protein (eGFP) and firefly luciferase (Luc), the eGFP–Luc fusion gene was removed from pEGFPLuc vector (BD Clontech laboratories, Palo Alto, CA, USA) with Eco47III and HpaI and ligated into the EcoRV site of pEF1/myc-HisB (InVitrogen, Carlsbad, CA, USA). The fragment of EF1α and EF-GL was excised with NruI and NotI and ligated into the Eco47III and XbaI sites of the lentiviral vector FUGW (a generous gift from Dr. D. Baltimore, California Institute of Technology) to produce F/EFGL, which constitutively expresses both eGFP and luciferase driven by the EF1α promoter. Recombinant lentiviruses were produced by transient transfection of the 293 T cells with the transducing vector and two packaging vectors: pVSVg, a plasmid expressing the VSVg envelope gene, and pΔ8.9, a plasmid expressing the HIV-1 gag/pol gene. The supernatants were collected after 48 h transfection. Multiple myeloma cell lines MM.1S, RPMI-8226, and CAG were transduced with the virus supernatant in the presence of 8 μg/ml polybrene, and transduced cells were identified by expression of eGFP and sorted by FACS.
Establishment of subcutaneous and disseminated myeloma xenografts and therapy
Nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (Jackson Laboratories, Bar Harbor, ME, USA) were housed and maintained in laminar flow cabinets under specific pathogen-free conditions in facilities under an institute-approved animal protocol. For the subcutaneous xenograft MM RPMI8226 mouse model, 10 to 12-week-old female mice were sublethally irradiated with 3 cGy from a Cesium γ-radiation source and inoculated with 10 × 106 tumor cells in the right flank. When tumor volumes approached 100 mm3, mice were divided into experimental cohorts of 8 to 10 mice each. Intraperitoneal (i.p.) injections of A12 antibody (40 mg/kg, three times per week) were administered for a total of 4 weeks. Control mice were treated with human immunoglobulin with the same protocol. For a second subcutaneous CAG mouse model, intraperitoneal injections of PBS or A12 alone, and bortezomib (0.25 mg/kg, iv, three times per week), and combinations were also given for a total of 4 weeks. Mice weight and tumor measurements were obtained three times weekly. Tumor volume was calculated using the formula mm3 = 4/3r 3, where r = (l + w)/4 using the two largest perpendicular axes of the tumor (l indicates length; w, width). Both lines were transduced with the eGFP–luc fusion gene to alternatively assess tumor burden by bioluminescence imaging. Mice were sacrificed when moribund or when one-dimensional tumor diameter exceeded 2.0 cm (according to the guidelines of the animal core facilities). Xenograft experiments were repeated at least twice. For establishing a disseminated xenograft NOD/SCID mouse model, 10 × 106 MM.1S cells stably expressing the eGFP–luciferase fusion protein were injected intravenously via tail vein. The tumor distribution was followed by serial whole-body noninvasive imaging of visible light emitted by luciferase-expressing MM cells upon injection of mice with luciferin. After day 9 post-tumor injection, a group of NOD/SCID mice with established disseminated MM lesion was divided into six cohorts, with statistically equivalent tumor burden between cohorts (as evaluated by bioluminescence imaging). Cohorts were treated with A12 alone (12 injections of 40 mg/kg each, i.p. three times per week), melphalan alone (single injection of 5 mg/kg each, i.p.), bortezomib alone (6 injections of 0.5 mg/kg each, i.v., 2 times per week), A12 plus melphalan, and A12 plus bortezomib, versus human immunoglubin-treated control mice.
Bioluminescence imaging (BLI)
Mice were given retro-obital injections of 75 mg/kg D-luciferin (Xenogen) in PBS, and bioluminescence imaging with a charge-coupled device (CCD) camera (IVIS, Xenogen, Alameda, CA, USA) was initiated 2 min after injection of luciferin. Bioluminescence images were obtained with a 15-cm field of view, binning (resolution) factor of 8, 1/f stop, and open filter. Imaging times were 1 to 60 s, depending on the quantum of luciferase activity. Bioluminescence from the region of interest (ROI) was quantified by Living Image R 2.20 software (Igor pro 4.06A, WaveMetrics, Inc, OR, USA) and Paint Shop Pro (Version 5.03, Jasc Software, Inc), and data were expressed as photon emission (photons/s/cm2/steradian). Background photon emission was defined from a ROI drawn over a healthy mouse with injection of luciferin.
Immunohistochemical staining of tumor xenograft tissues
Tumor-bearing mice were sacrificed at the end of treatment. Subcutaneous RPMI8226 MM xenografts recovered at necropsy were evaluated by immunohistochemistry after fixation in 10% formalin-and-paraffin embedding. Four-micron-thick sections were stained with hematoxylin and eosin (H&E) and Giemsa for histologic examination. Tumor microvessel density (MVD) was evaluated by immunohistochemical staining of tissues using antibodies against mouse endothelial cells CD31 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and Flk-1 (1:200; PharMingen, San Diego, CA, USA). For determination of CD31 expression, the whole tumor tissue section was photographed using MetaMorph (Molecular Devices Corporation, Downingtown, PA, USA) Zeiss Axiovert 200 M (Carl Zeiss, Inc.) imaging system. Then positive pixels (CD31) and whole section pixels were quantified using MetaMorph Offline software followed by quantification of positive pixels and the total section pixels (MetaMorph Offline software, Version 6.2r6, Molecular Devices Corp., PA, USA). The results were expressed as percentage of positive pixels versus total section pixels. Evaluation of tissue proliferation rates was performed with the monoclonal NCL-Ki67p antibody (1:200; Novocastra Laboratories Ltd., UK), and mitotic rates with the polyclonal anti-phospho-Histone H3 antibody (1:200; Upstate Biotechnology, NY, USA) using antigen retrieval. The number of Ki-67 and Phospho-Histone H3 positive cells was assessed in three areas containing the highest positive cell expression using a Zeiss Axioplan 2 light microscope. Apoptotic cells were identified by use of standard terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay.
Production of human VEGF in tumor cells under serum starvation conditions
Human vascular endothelial growth factor (VEGF) levels in MM culture supernatants were measured by a quantitative sandwich enzyme-linked immunoassay technique (ELISA) according to the manufacturer’s instructions (Human VEGF Immunoassay, R&D systems, MN, USA). The MM cells were incubated in a 24 well plate (1∼2 × 105/1 ml) with serum-free RPMI 1640 medium for 24 h. The supernatant was collected and subjected to human VEGF ELISA assay with intensity of the color measured on an ELISA reader (Thermo Max, Molecular Devices, CA, USA) at 450 nm with a reference wavelength to 650 nm. The results were then plotted against the standard curve to provide the actual concentrations. Samples were plated in triplicate wells and experiments were repeated three times.
Statistical analysis
All analyses were done using Stata 7.0 statistical software (Stata Corporation, College Station, TX, USA), with P < 0.05 considered to be significant. For comparing tumor-associated parameters, the nonpaired Student’s t test was used. Log–rank tests were used to calculate the statistical significance of difference of Kaplan–Meier survival curves. The overall survival of mice was defined as the time between i.v. injection of tumor cells and sacrifice or death, and was compared across different treatment groups with Kaplan–Meier survival analysis.
Results
Expression of IGF-IR on the surface of human MM cell lines
For systemically administered anti-IGF-IR antibodies to inhibit malignant growth, tumor cells must express surface IGF-IR accessible for antibody binding. Flow cytometry analysis of a panel of human MM cell lines was performed either using anti-IGF-IR antibody conjugated to fluorescent dyes or by the sandwich methods using antibody A12 against IGF-IR. As shown in Fig. 1, all myeloma cell lines screened expressed IGF-IR on the cell surface, but relative expression level varied considerably between cell lines. However, if results were expressed as the mean fluorescence intensity ratio indicated in Fig. 1, the expression of IGF-IR is rather homogeneous except for a low expresser in CAG line and a high in U266 line.

Screening of IGF-IR expression on human MM cell lines by flow cytometric analysis. Results are representative of the lines tested and of analyses with two different anti-IGF-IR-specific monoclonal antibodies (R&D systems anti-IGF-IR-FITC and ImClone A12). Both antibodies generated comparable results for IGF-IR expression. White peaks correspond to isotype-matched control, and shaded peaks as IGF-IR. The number in the histograms represents the mean fluorescence intensity ratio of positive versus isotype control
Inhibition of IGF-IR signaling by A12
Monoclonal antibody A12 is a selective antagonist of the human IGF-IR. It has been previously shown that the A12 could inhibit IGF-I-dependent mitogenesis of RPMI8226 tumor cells in vitro [32]. Here, we sought to assess receptor and downstream effector molecule phosphorylation to determine the extent to which ligand-dependent signaling was inhibited in MM cells by A12. As shown in Fig. 2a, the A12 significantly inhibited tyrosine phosphorylation of the IGF-IR in response to exogenous IGF-I. Furthermore, activation of downstream signaling pathways was also suppressed, as demonstrated by inhibition of ligand-dependent phosphorylation of Akt and ERK1, 2 (Fig. 2a). The A12 has further been shown to suppress IGF-IR signaling by effecting rapid internalization and lysosomal-mediated receptor degradation following specific binding to surface IGF-IR [32]. RPMI8226 cells were treated with either A12 or IGF-I for up to 24 h. As shown in Fig. 2b, the A12 treatment caused a significant reduction in total cellular IGF-IR within 1 h, reaching 70% reduction of total cellular IGF-IR levels for the duration of analysis. In contrast, exposure to ligand did not alter the steady-state levels of receptor. Exposure to A12 therefore inhibited IGF-IR signaling by blocking the binding of ligand to the receptor and, further, by effecting a reduction in receptor density within the tumor cells.

Antibody-mediated inhibition of ligand-dependent receptor auto-phosphorylation and degradation. a RPMI8226 cells were pretreated with A12 (100 nM) or control antibody (100 nM anti-KLH) prior to addition of IGF-I (10 nM). Total receptor was immunoprecipitated (IP) from total cell lysate and a western blot of the IGF-IR IP probed for tyrosine phosphorylation. For detection of phosphorylated downstream signaling molecules, western blots of total lysate were probed for phospho-Akt and phospho-MAPK (pERK1, 2), then stripped and reprobed for total Akt and ERK 1, 2. Band intensity was determined by densitometric analysis using an Alphaimager (AlphaInnotech), normalized to total protein, and plotted relative to control values (100%) b RPMI8226 cells were treated with A12 (100 nM) or IGF-I (100 nM) in culture and lysates generated at various time points post-treatment. IGF-IR was detected by western blot for assessment of antibody-mediated receptor degradation
A12 inhibition of serum or IGF-I stimulated MM cell growth in vitro
Expression of the IGF-IR was identified in many MM cell lines (Fig. 1). It was, therefore, important to determine whether this activation was biologically significant in terms of tumor cell growth. To address this question, we analyzed the proliferative effect of IGF-I on MM cell growth in vitro by XTT tetrazonium assay. Compared to controls, IGF-I at 50 ng/ml stimulated a twofold to 3.5-fold increase in viable cells recovered in 4 of 6 lines tested (Fig. 3a), with the greatest response seen with H929 and OPM-2 cells which had an intermediate level of surface IGF-IR expression (Fig. 1).

Effect of IGF-I and antibody A12 on in vitro proliferation of MM cells. a Effect of IGF-I on in vitro proliferation of MM cells. After serum starvation for 16 h, cells from indicated lines were plated at 5 × 103/well in 96 well plates in the presence (solid bar) or absence (open bar) of 50 ng/ml of IGF-I for 52 h in the absence of serum. The cell proliferation was measured by XTT tetrazonium assay using a 20-h incubation with XTT salt and PMS. IGF-I-induced cell proliferation is presented as the fold increase calculated by dividing the mean value of the untreated wells by that of IGF-I-stimulated counterpart. Data represent average values of triplicate samples from three separated experiments. b In vitro sequential quantification of H929 cell survival after specific inhibition of serum-triggered IGF-IR activation by XTT tetrazonium assay. After serum starvation for 16 h, myeloma H929 cells were plated at 5 × 103/well in 96 well plates in the absence (open bar), presence of 2% FCS (black bar), and 2%FCS plus 200 nM A12 (grey bar) for 4, 28 and 52 h, followed by XTT assay as described previously. Data represent average values of triplicate samples from three separate experiments (*P < 0.01). c In vitro quantification of myeloma cell survival after specific inhibition of IGF-I triggered IGF-IR activation in the XTT tetrazonium assay. After serum starvation for 16 h, cells from indicated lines were plated at 5 × 103/well in 96 well plates in the absence (open bar), presence of 10 ng/ml IGF-I (black bar), and 10 ng/ml IGF-I plus 200 nM A12 (grey bar) for 52 h, followed by XTT assay as described previously. Data represent average values of triplicate samples from three separate experiments (#P < 0.01)
To evaluate the degree to which inhibition of IGF-IR function can suppress the ability of serum (which contains IGFs) to stimulate an increase in viable MM cells, we undertook in vitro sequential quantitation of H929 cell viability after specific inhibition of serum-induced IGF-IR activation by A12. After serum starvation for 16 h, H929 cells were plated in the absence or presence of 2% FCS alone, or 2% FCS plus 200 nM A12 for 24, 48 and 72 h, followed by XTT assay as described previously. Typically, cells were incubated first with A12 for about 30 min to 2 h and then treated with serum. After 3 days of growth, A12 treatment caused up to 60% inhibition of the serum-stimulated proliferation and survival of H929 cells, implicating the essential role of IGF-I as an MM growth factor present in serum (Fig. 3b, P < 0.01). The A12 inhibited the serum-stimulated proliferation of H929 cell line in the presence of several different batches of serum and over a range of concentrations (1.25–6% FCS; data not shown).
We then explored if cells that were stimulated with IGF-I could also be inhibited in their proliferation and survival by co-incubation with the blocking antibody A12. Recombinant IGF-I stimulated proliferation of a number of MM cell lines following 16 h of serum deprivation, and this effect was almost completely inhibited in the presence of A12 (Fig. 3c).
Antitumor activity of A12 in subcutaneous xenograft MM tumor models
The A12 has exhibited strong inhibitory activity on IGF-dependent mitogenesis and proliferation of MM cells in vitro. To assess its activity on MM tumor growth in vivo, subcutaneous xenograft tumor models in NOD/SCID mice were established. Two cell lines, RPMI8226 and CAG, were selected for tumor inhibition study. Dose–response analysis of A12 has been performed previously in the MCF7 breast cancer model using doses of 0.01, 0.1, and 1 mg at 3-day intervals, showing that 1 mg per mouse (40 mg/kg) was the optimal therapeutic dosage for treatment [32]. The 40 mg/kg dose and schedule was therefore employed throughout these studies. To evaluate the combination effects of A12 with other anti-myeloma agents, we included Bortezomib, a proteasome inhibitor in the study. As shown in Fig. 4a, b, the animals treated with A12 alone exhibited inhibition of RPMI8226 tumor cell growth. The tumor growth measured by tumor volume showed that single-agent A12 treatment produced > 60% inhibition of RPMI8226 tumor growth (P = 0.025, day 27, A12 vs. control) but less (∼30%) inhibition of tumor growth in the CAG tumor model, a MM line with reduced expression of IGF-IR (Fig. 4a, c). In the CAG model, single agent A12 controlled tumor growth as effectively as the proteasome inhibitor bortezomib (P < 0.05, day 27, single agent vs. control), but there was no enhanced benefit when the drugs were given in combination (Fig. 4c). However, quantitation of tumor burden by BLI, which compared favorably to tumor diameter measurement, suggested a modest benefit of combination therapy, and may be a potentially more sensitive method for determining tumor burden (Fig. 4d). In none of the studies was any obviously treatment-related toxicity or change in body weight noted when compared to the saline-treated control animals. Wherein the anti-tumor effect of A12 on MM can be attributed to its effect on inhibiting signaling via the IGF-IR, because it is an IgG1 antibody, the possible contribution of immune effector functions, including ADCC, to the anti-tumor activity of this antibody cannot be excluded.

Anti-tumor activities of human anti-IGF-IR monoclonal antibody (A12) or in combination with other anti-myeloma agents in subcutaneous NOD/SCID MM mouse models. a Suppression of growth of RPMI8226 MM xenografts in a subcutaneous mouse model by antibody A12. Established RPMI8226 xenografts (100 mm3) were treated with A12. The A12 was administered intraperitonealy (i.p., 40 mg/kg per mouse) three times a week for a total of 4 weeks of treatment. Control mice were treated with human immunoglobulin using the same protocol as A12. Data plotted are mean ± SEM for tumor volume. b Quantification of dorsal tumor burden of RPMI8226 model by bioluminescence imaging at day 28. Data plotted are mean ± SEM for photon emission (P < 0.05). c Suppression of growth of CAG MM xenografts in subcutaneous mouse model by A12 alone, bortezomib alone, or in combination of both agents. Established CAG xenografts (100 mm3) were treated with A12 (12 injections of 40 mg/kg each, i.p. three times per week), bortezomib alone (12 injections of 0.25 mg/kg each, i.v., three times per week), or in combination. Control mice were treated with human immunoglobulin using the same protocol as A12. Data plotted are mean ± SEM for tumor volume. d Quantification of dorsal tumor burden of CAG model by bioluminescence imaging at day 28. Data plotted are mean ± SEM for photon emission (compared to control, P > 0.05 for A12 alone; P < 0.05 for bortezomib alone; P < 0.01 for combination)
A12 inhibition of myeloma cell VEGF production
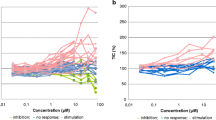
The data presented earlier demonstrate the existence of active IGF-I signaling pathways in most MM cells, and that suppression of IGF-I/IGF-IR signaling can effectively inhibit tumor growth both in vitro and in vivo. We have previously seen a marked increase in apoptotic tumor cells in A12-treated subcutaneous breast and prostate cancer mouse models [32, 33]. To address this in MM tumors, we analyzed subcutaneous RPMI8226 tumor tissues at the end of A12 treatment. Tumor specimens from representative animals were resected, fixed and processed for immunohistochemical staining. In this model, treatment with A12 effected a marked reduction in the number of proliferating cells (Fig. 5a) as demonstrated by a reduction in Ki-67 and phospho-Histone H3 positive tumor cells. However, TUNEL analysis of tumor tissue did not reflect an increase in apoptosis due to A12 treatment, but rather significant necrosis, as evidenced by large acellular regions throughout the tumor tissue (Fig. 5a). Strikingly, the A12 treated tumor had significantly decreased vascularization of up to tenfold compared to control tumors as determined by anti-mouse CD31 staining of murine endothelium (Fig. 5b). Since human VEGF can trigger mouse angiogenesis, we evaluated VEGF production by the MM lines using ELISA assay. We determined that most MM lines secreted a significant quantum of VEGF (Fig. 6a), and further that the A12 could prevent IGF-mediated induction of VEGF secretion in MM cells in vitro (Fig. 6b). Interestingly, upon challenge with A12 in serum-free medium, constitutive secretion of VEGF from MM cells was markedly reduced by as little as 67 nM antibody (Fig. 6c, d). This effect may be due to suppression of autocrine stimulation of IGF-IR signaling or perhaps due to feedback from IGF-IR receptor down-modulation in treated cells. These data collectively indicate that treatment of myeloma cells in culture can suppress VEGF production, and suggest a putative anti-angiogenic mechanism associated with IGF-I receptor antibody antagonism.

Histological analysis of tumor sections from antibody A12 treated RPMI8226 xenografts in a subcutaneous myeloma model. a After termination of the treatment, tumor specimens from representative animals were resected, fixed and processed for immunohistochemical staining. Antibodies against nuclear antigen Ki-67 for proliferating cells, and phospho-Histone-H3 for dividing cells were used. Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay was performed for cell apoptosis examination (magnification 20×). B. The whole tumor sections stained with anti-mouse CD31/PECAM-1 for identification of mouse endothelial cells were photographed using MetaMorph Zeiss Axiovert 200 M (Inverted Stand) imaging system. The positive pixels (CD31) and the whole section pixels were quantified using MetaMorph Offline software. At least three sections were evaluated in each specimen (magnification 60×). The result was expressed as the average percentage of positive pixels versus the total section pixels (P < 0.001 for A12 vs control)

Human VEGF secretion by MM cells in vitro and impact of IGF-IR inhibition by antibody A12 on MM cells VEGF production. a Autocrine production of VEGF in human MM cells under serum-free conditions. After serum starvation for 16 h, cells from indicated lines were plated at 2 × 105/well in 24 well plates in the absence of serum and cultured for 24 h. Supernatant was harvested and subjected to VEGF165 assay by a commercial available ELISA kit. The data are the average of two independent results. b IGF-IR inhibition by A12 suppresses IGF-I stimulated VEGF production by MM cells. Serum starved MM cells as indicated were incubated in the presence of IGF-I (10 ng/ml) alone or IGF-I plus A12 (200 nM) in either QBSF or RPMI1640 medium for 24 h. Supernatant was harvested and subjected to VEGF165 assay. c, d IGF-IR inhibition by A12 suppresses the constitutive secretion of VEGF by MM cells. Serum starved H929 and MM.1S cells were cultured with A12 at a concentration of 67, 100 and 200 nM for 24 h. Supernatant was harvested and subjected to VEGF165 assay. The viability of MM cells was assessed by trypan blue staining
Combination A12 with anti-myeloma agents in a disseminated xenograft tumor model
The in vivo preclinical testing of investigational therapies for MM is hampered by the fact that most human MM tumor xenograft mouse models fail to recapitulate the extensive marrow tumor infiltration and development of diffuse skeletal lesions of the human disease even after intravenously (i.v.) injections of tumor cells. Furthermore, it has proven difficult to monitor the exact site(s) of tumor growth or reliably quantify disseminated tumor mass. We therefore utilized an in vivo MM model in NOD/SCID mice in which diffuse MM lesions developed after tail vein i.v. injection of human MM.1S cells stably transfected with eGFP–Luciferase. This system enables whole-body real-time BLI to detect luciferase positive MM cells and monitor the development and progression of disseminated MM disease [38]. We used this in vivo model to test the biological activity of A12 therapy on disseminated MM disease alone and in combination with conventional therapeutic agents for MM. Mice (n = 10 per group) were treated with A12 alone, a single dose of melphalan alone (that was half the dose that produced > 95% tumor regression of subcutaneous tumor), or a combination of the two agents. We also treated mice with bortezomib alone, or bortezomib plus A12. At the end of 4 weeks treatment, the tumor burden in A12 (Fig. 7a) was qualitatively reduced in comparison to control, but the change total body burden was not statistically significant (Fig. 7b, Table 1). However, tumor burden in the combination of A12 and melphalan was significant in comparison to control or either single agent treatment at 2 weeks, and exhibited a trend for continued reduced tumor burden at 4 weeks (Table 1). Combination treatment of A12 with the proteasome inhibitor bortezomib also trended lower than single agent, but was not statistically significant in comparison to single agent bortezomib. Notably, however, although overall survival was significantly prolonged in each single agent treatment group relative to control, the combination groups showed significantly greater survival than single agent alone, including single agent bortezomib. This demonstrated that inclusion of anti-IGF-IR antibody with conventional therapy could improve survival in a disseminated MM model (Fig. 7c, P < 0.001). There was no difference in body weight between treatment groups (P = 0.95) at 30 days and no other significant treatment-related toxicity discernible by necropsy and histopathological evaluation (data not shown). Taken together, these results demonstrate that abrogation of IGF-I/IGF-IR signaling after cytotoxic therapy may enhance the efficacy of therapies that are currently used in MM treatment.

Anti-tumor activity of A12 in a disseminated MM xenograft model and quantification of tumor burden by bioluminescence imaging. A disseminated myeloma xenograft model was established in NOD/SCID mouse using intravenous injection of transduced MM.1S cells. Tumor distribution was followed by serial whole-body noninvasive imaging of visible light emitted by luciferase-expressing MM cells upon injection of mice with luciferin. Dorsal and ventral images were obtained at day 8 (week 2), 23 (week 4), 37, and 43. a Week 2 images from control and single agent A12 groups, representative of MM tumor cell distribution in vivo and inhibitory effect of anti-IGF-IR therapy in this model. b Quantitation of dorsal photon emission in treatment groups to evaluate tumor burden. Photon emission was determined from images obtained on week 0, 2 and 4 for all mice in each cohort and mean +/− SEM plotted. Accompanying statistical analysis for each group is listed in Table 1. Ventral image analysis for each treatment group produced similar results. c Kaplan–Meier survival curve of single agent and combination therapy in the disseminated MM model. A12 enhanced the in vivo antitumor effect of melphalan and bortezomib, as shown by the prolongation of overall survival of mice receiving the combination of the two agents (log-rank test, *P < 0.02 for A12 alone treated cohort versus control; **P < 0.005 for A12 + melphalan and A12 + bortezomib treated cohorts versus single drug alone treated cohorts)
Discussion
The high-affinity fully human monoclonal antibody, A12, has been shown to be a potent anti-tumor agent in various xenograft models, including MM. The ubiquitous expression of the IGF-IR receptor on MM cells (Fig. 1), and the production of IGF ligands by stromal cells, osteoblasts and endothelial cells in the bone marrow [34] suggests a role for IGF as a paracrine growth stimulus for tumors. The IGFs are frequently overexpressed in human tumors, often in concert with IGF-IR overexpression, to generate autocrine activation of the IGF-IR signaling pathway [35]. Blocking autocrine or paracrine signaling by both ligands is therefore likely to be necessary for an antagonist antibody to be an effective therapeutic approach.
In this study, we have focused on MM, as a disease that may be significantly dependent on IGF-IR function [36]. We demonstrated an in vivo antitumor activity of the IGF-IR antibody A12 when used as a single agent, or when combined with melphalan cytotoxic chemotherapy or with bortezomib proteasome inhibitor therapy in both subcutaneous and disseminated mouse xenograft models without significant treatment-related toxicity. That combination therapy exhibited more effective antitumor activity is particular of interest. Yee et al. [37] reported that when scFv–Fc (a chimeric humanized single chain antibody against IGF-IR) combined with tamoxifen, a selective estrogen receptor modulator could significantly enhance suppression of tumor growth in a T61 human breast cancer model. Recently, Mitsiades et al. [38] found that selective IGF-IR tyrosine kinase inhibitor NVP-ADW742 could increase the sensitivity of MM cells to various cytotoxic chemotherapeutic agents in vitro. Our combination strategies with anti-IGF-IR antibody therapy in mice with either subcutaneous or disseminated MM xenografts exhibited enhanced antitumor activity and prolonged overall survival, providing a framework for the potential clinical applications of targeting IGF-IR to enhance the efficacy of conventional cytotoxic drug or proteasome inhibitor by blocking key survival signals (Fig. 1).
The IGF-IR signaling pathway has been implicated as a causative factor in the development of mouse plasmacytomas, with tumors showing up-regulated IGF-IR expression and exhibiting a mitogenic response to IGF-I [39]. Plasmacytoma induction by a retrovirus containing oncogene raf/myc was associated with either constitutively or IGF-I-dependent activation of the insulin receptor substrate 2/phosphatidylinositol 3′-kinase/p70S6K pathway [40]. Furthermore, expression of a dominant-negative mutant of IGF-IR in plasmacytoma lines strongly suppressed tumorigenesis in vivo [39].
In addition to the established functions of the IGF-IR listed above, our study also highlighted its role in modulating tumor angiogenesis. Reinmuth et al. have reported that IGF-I increases VEGF expression in human colon carcinoma cells [41]. This observation is of particular interest because the sites of colon cancer growth express IGF-I constitutively. The organ with the highest levels of IGF-I, the liver [42], is also the primary organ of colon cancer metastasis. In addition to a paracrine effect, IGF-I and IGF-II may also act in an autocrine manner since colon mucosa and colon cancers themselves synthesize significant quantities of IGF-I [43]. In a spontaneous MM mouse model, the MM cells were shown to produce large quantities of VEGF in vitro after IGF-I stimulation [44]. This tumor response appeared to involve activation of the MEK/ERK pathway since it could be suppressed by addition of the MEK inhibitor PD98059. Clinically, marked elevation of tumor microvessel density (MVD) is seen in MM bone marrow biopsies and the greater the MVD, the poorer the prognosis [45]. Tumor-associated angiogenesis as reflected by MVD is a biologically relevant and important prognostic feature that can be targeted by antiangiogeneic therapy, such as thalidomide, which has remarkable antitumor activity in advanced and refractory MM [46]. To evaluate the importance of IGF-IR in human MM and its contribution to tumor angiogenesis, we investigated autocrine VEGF production by MM cells. We found that all myeloma cells secrete significant quantum of VEGF (up to 900 pg/ml/2 × 105 cells) under serum starvation condition in vitro. Specific inhibition of IGF-IR signaling by A12 can sharply decrease VEGF production by MM cells, suggesting that IGF-IR signaling is involved in tumor production of VEGF. This in turn facilitates tumor angiogenesis by a paracrine stimulation of locally invasive microvessels or recruitment of endothelial progenitors from the bone marrow [47, 48]. Furthermore, we assessed MVD in whole tumor section of subcutaneous xenografts of RPMI8226 tumor following immunohistochemical staining for mouse endothelial markers CD31 and Flk-1. We showed a marked decrease in tumor staining for these markers in the A12 treatment group compared with controls, indicating that inhibition of tumor growth by A12 was associated with a profound reduction in tumor vascularization. With a reduced blood supply, tumor growth was inhibited in association with greatly reduced mitotic activity and increased tumor cell necrosis. Suppression of tumor VEGF production may also inhibit tumor growth via disruption of an autocrine pathway involving tumor expression of VEGF and functional VEGF receptors has been demonstrated in MM [49–51].
The studies we have reported are novel because they are the first to address the role of IGF-IR on tumor angiogenesis in human MM both in vitro and in vivo. The contribution of IGF-I-induced angiogenesis may be even more important within the bone marrow which provides a critical niche where MM cells interact with IGF-I-expressing stroma, osteoblasts and microvascular endothelium [52]. We postulate that when IGF-I stimulates VEGF secretion by the MM cells in the bone marrow, it results in paracrine stimulation of endothelial cell growth and recruitment with enhanced vascularization as well as an autocrine stimulation of tumor growth. In addition, since IGF-IR expression on MM cells is up-regulated after their contact with BM endothelial cells [53] a vicious cycle is established in which increased IGF-IR signaling increases VEGF production which in turn increases tumor autocrine proliferation and vascularization. This in turn induces more IGF-IR expression and IGF-ligand production.
In conclusion, our data demonstrate the critical role of IGF-IR function in MM cells at various stages in tumor growth both in vitro and in vivo. Specifically, this report is the first to demonstrate that IGF-I can induce VEGF secretion in human MM cells, and inhibition of IGF-IR function may be an indirect means of inhibiting VEGF expression and angiogenesis. We also provide evidence that anti-IGF-IR antibody-mediated tumor inhibition is more effective when combined with either melphalan chemotherapy or bortezomib-mediated proteasome inhibition—two agents that have been shown to be particularly effective in MM therapy.
References
Kyle RA, Rajkumar SV (2004) Multiple myeloma. N Engl J Med 351:1860
Mitsiades CS, Mitsiades N, Munshi NC, Anderson KC (2004) Focus on multiple myeloma. Cancer Cell 6:439
Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, Walker R, Crowley J (2004) Treatment of multiple myeloma. Blood 103:20
Fassas A, Shaughnessy J, Barlogie B (2005) Cure of myeloma: hype or reality? Bone Marrow Transplant 35:215
Hideshima T, Chauhan D, Ishitsuka K, Yasui H, Raje N, Kumar S, Podar K, Mitsiades C, Hideshima H, Bonham L, Munshi NC, Richardson PG, Singer JW, Anderson KC (2005) Molecular characterization of PS-341 (bortezomib) resistance: implications for overcoming resistance using lysophosphatidic acid acyltransferase (LPAAT)-beta inhibitors. Oncogene 24:3121
Pollak MN, Schernhammer ES, Hankinson SE (2004) Insulin-like growth factors and neoplasia. Nat Rev Cancer 4:505
Weber MM, Fottner C, Liu SB, Jung MC, Engelhardt D, Baretton GB (2002) Overexpression of the insulin-like growth factor I receptor in human colon carcinomas. Cancer 95:2086
Hellawell GO, Turner GD, Davies DR, Poulsom R, Brewster SF, Macaulay VM (2002) Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res 62:2942
Happerfield LC, Miles DW, Barnes DM, Thomsen LL, Smith P, Hanby A (1997) The localization of the insulin-like growth factor receptor I (IGFR-I) in benign and malignant breast tissue. J Pathol 183:412
Muller M, Dietel M, Turzynski A, Wiechen K (1998) Antisense phosphorothioate oligodeoxynucleotide down-regulation of the insulin-like growth factor I receptor in ovarian cancer cells. Int J Cancer 77:567
Bergmann U, Funatomi H, Yokoyama M, Beger HG, Korc M (1995) Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res 55:2007
Cullen KJ, Yee D, Sly WS, Perdue J, Hampton B, Lippman ME, Rosen N (1990) Insulin-like growth factor receptor expression and function in human breast cancer. Cancer Res 50:48
Ankrapp DP, Bevan DR (1993) Insulin-like growth factor-I and human lung fibroblast-derived insulin-like growth factor I stimulate the proliferation of human lung carcinoma cells in vitro. Cancer Res 53:3399
Guo YS, Jin GF, Townsend CM Jr, Zhang T, Sheng HM, Beauchamp RD, Thompson JC (1995) Insulin-like growth factor II expression in carcinoma in colon cell lines: implications for autocrine actions. J Am Coll Surg 181:145
Kappel CC, Velez-Yanguas MC, Hirschfeld S, Helman LJ (1994) Human osteosarcoma cell lines are dependent on insulin-like growth factor I for in vitro growth. Cancer Res 54:2803
Steller MA, Delgado CH, Bartels CJ, Woodworth CD, Zou Z (1996) Overexpression of the insulin-like growth factor I receptor and autocrine stimulation in human cervical cancer cells. Cancer Res 56:1761
Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M (1998) Plasma insulin-like growth factor I and prostate cancer risk: a prospective study. Science 279:563
Ma J, Pollak MN, Giovannucci E, Chan JM, Tao Y, Hennekens CH, Stampfer MJ (1999) Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst 91:620
Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X (1999) Plasma levels of insulin-like growth factor I and lung cancer risk: a case–control analysis. J Natl Cancer Inst 91:151
Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, Rosner B, Speizer FE, Pollak M (1998) Circulating concentrations of insulin-like growth factor I and risk of breast cancer. Lancet 351:1393
Baserga R, Hongo A, Rubini M, Prisco M, Valentinis B (1997) The IGF-I receptor in cell growth, transformation and apoptosis. Biochim Biophys Acta 1332:F105
Navarro M, Baserga R (2001) Limited redundancy of survival signals from the type 1 insulin-like growth factor receptor. Endocrinology 142:1073
Freund GG, Kulas DT, Way BA, Mooney RA (1994) Functional insulin and insulin-like growth factor-1 receptors are preferentially expressed in multiple myeloma cell lines as compared to B-lymphoblastoid cell lines. Cancer Res 54:3179
Ge NL, Rudikoff S (2000) Insulin-like growth factor I is a dual effector of multiple myeloma cell growth. Blood 96:2856
Carroll M, Abrams CS (2004) Signaling, drugs and apoptosis of myeloma cells. Cancer Biol Ther 3:195
Abroun S, Ishikawa H, Tsuyama N, Liu S, Li FJ, Otsuyama K, Zheng X, Obata M, Kawano MM (2004) Receptor synergy of interleukin-6 (IL-6) and insulin-like growth factor-I that highly express IL-6 receptor alpha myeloma cells. Blood 103:2291
Tucci A, Bonadonna S, Cattaneo C, Ungari M, Giustina A, Guiseppe R (2003) Transformation of a MGUS to overt multiple myeloma: the possible role of a pituitary macroadenoma secreting high levels of insulin-like growth factor 1 (IGF-1). Leuk Lymphoma 44:543
Qiang YW, Kopantzev E, Rudikoff S (2002) Insulin like growth factor-I signaling in multiple myeloma: downstream elements, functional correlates, and pathway cross-talk. Blood 99:4138
Zhang X, Yee D (2000) Tyrosine kinase signalling in breast cancer: insulin-like growth factors and their receptors in breast cancer. Breast Cancer Res 2:170
Tai YT, Podar K, Catley L, Tseng YH, Akiyama M, Shringarpure R, Burger R, Hideshima T, Chauhan D, Mitsiades N, Richardson P, Munshi NC, Kahn CR, Mitsiades C, Anderson KC (2003) Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3′-kinase/AKT signaling. Cancer Res 63:5850
Qiang YW, Yao L, Tosato G, Rudikoff S (2004) Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood 103:301
Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, Bassi R, Abdullah R, Hooper AT, Koo H, Jimenez X, Johnson D, Apblett R, Kussie P, Bohlen P, Witte L, Hicklin DJ, Ludwig DL (2003) A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res 63:8912
Wu JD, Odman A, Higgins LM, Haugk K, Vessella R, Ludwig DL, Plymate SR (2005) In vivo effects of the human type 1 insulin-like growth factor receptor antibody A12 on androgen-dependent and androgen-independent xenograft human prostate tumors. Clin Cancer Res 11:3065
Vanderkerken K, Asosingh K, Braet F, Van Riet I, Van Camp B (1999) Insulin-like growth factor-1 acts as a chemoattractant factor for 5T2 multiple myeloma cells. Blood 93:235
Gross JM, Yee D (2003) The type-1 insulin-like growth factor receptor tyrosine kinase and breast cancer: biology and therapeutic relevance. Cancer Metastasis Rev 22:327
Qiang YW, Yao L, Tosato G, Rudikoff S (2004) Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood 103:301
Ye JJ, Liang SJ, Guo N, Li SL, Wu AM, Giannini S, Sachdev D, Yee D, Brunner N, Ikle D, Fujita-Yamaguchi Y (2003) Combined effects of tamoxifen and a chimeric humanized single chain antibody against the type I IGF receptor on breast tumor growth in vivo. Horm Metab Res 35:836
Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringapure R, Akiyama M, Hideshima T, Chauhan D, Joseph M, Libermann TA, Garcia-Echeverria C, Pearson MA, Hofmann F, Anderson KC, Kung AL (2004) Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 5:221
Li W, Hyun T, Heller M, Yam A, Flechner L, Pierce JH, Rudikoff S (2000) Activation of insulin-like growth factor I receptor signaling pathway is critical for mouse plasma cell tumor growth. Cancer Res 60:3909
Hilbert DM, Pumphrey JG, Troppmair J, Rapp UR, Rudikoff S (1993) Susceptibility and resistance to J3V1 retrovirus-induced murine plasmacytomagenesis in reconstituted severe combined immunodeficient mice. Oncogene 8:1993
Reinmuth N, Liu W, Fan F, Jung YD, Ahmad SA, Stoeltzing O, Bucana CD, Radinsky R, Ellis LM (2002) Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer. Clin Cancer Res 8:3259
Jones JI, Clemmons DR (1995) Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev 16:3
Hakam A, Yeatman TJ, Lu L, Mora L, Marcet G, Nicosia SV, Karl RC, Coppola D (1999) Expression of insulin-like growth factor-1 receptor in human colorectal cancer. Hum Pathol 30:1128
Menu E, Kooijman R, Van Valckenborgh E, Asosingh K, Bakkus M, Van Camp B, Vanderkerken K (2004) Specific roles for the PI3K and the MEK-ERK pathway in IGF-1-stimulated chemotaxis, VEGF secretion and proliferation of multiple myeloma cells: study in the 5T33MM model. Br J Cancer 90:1076
Ribas C, Colleoni GW, Silva MR, Carregoza MJ, Bordin JO (2004) Prognostic significance of vascular endothelial growth factor immunoexpression in the context of adverse standard prognostic factors in multiple myeloma. Eur J Haematol 73:311
Barlogie B, Desikan R, Eddlemon P, Spencer T, Zeldis J, Munshi N, Badros A, Zangari M, Anaissie E, Epstein J, Shaughnessy J, Ayers D, Spoon D, Tricot G (2001) Extended survival in advanced and refractory multiple myeloma after single-agent thalidomide: identification of prognostic factors in a phase 2 study of 169 patients. Blood 98:492
Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, Hicklin DJ, Zhu Z, Bohlen P, Witte L, Hendrikx J, Hackett NR, Crystal RG, Moore MA, Werb Z, Lyden D, Rafii S (2002) Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med 8:841
Rafii S, Lyden D (2003) Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med 9:702
Ria R, Roccaro AM, Merchionne F, Vacca A, Dammacco F, Ribatti D (2003) Vascular endothelial growth factor and its receptors in multiple myeloma. Leukemia 17:1961
Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, Lin BK, Gupta D, Shima Y, Chauhan D, Mitsiades C, Raje N, Richardson P, Anderson KC (2001) Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 98:428
Kumar S, Witzig TE, Timm M, Haug J, Wellik L, Fonseca R, Greipp PR, Rajkumar SV (2003) Expression of VEGF and its receptors by myeloma cells. Leukemia 17:2025
Moschos SJ, Mantzoros CS (2002) The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology 63:317
Asosingh K, Radl J, Van Riet I, Van Camp B, Vanderkerken K (2000) The 5TMM series: a useful in vivo mouse model of human multiple myeloma. Hematol J 1:351
Acknowledgments
The authors would like to acknowledge Kang Zhang for excellent assistance with NOD-SCID mouse model, WeiHong Yang for performing the paraffin block and immunohistochemical staining on tumor sections, Katia Manova at the Molecular Cytology Core Facility for reviewing pathological slides, Pat Zanzonico at Animal Imaging Core for helping whole mouse bioimaging, and Kate deBeer for preparing the manuscript. M.A.S.M was supported by a SCOR Grant in Multiple Myeloma (Selina Chen-Kiang PI). This work was further supported by a translational grant from the LSA to M.A.S.M.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, KD., Zhou, L., Burtrum, D. et al. Antibody targeting of the insulin-like growth factor I receptor enhances the anti-tumor response of multiple myeloma to chemotherapy through inhibition of tumor proliferation and angiogenesis . Cancer Immunol Immunother 56, 343–357 (2007). https://doi.org/10.1007/s00262-006-0196-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-006-0196-9