
Abstract
Prostate cancer is the most commonly diagnosed form of cancer and the second leading cancer-related death among men in the Western civilization. Since no effective therapy exists for this tumor after progression beyond resectable boundaries, there is an urgent need for new treatment strategies. Prostate specific membrane antigen (PSMA) represents an excellent target on prostate cancer cells, and therefore specific immunotherapy may be a novel therapeutic option for the management of this tumor. We constructed a fully recombinant immunotoxin (A5-PE40) from a single-chain antibody fragment (scFv) against cell-adherent PSMA and a truncated form of Pseudomonas exotoxin A (PE40) lacking its natural binding domain Ia. The scFv A5 was obtained from a mAb elicited with native PSMA by phage display technology and direct selection on cells carrying the antigen. The bacterially expressed and purified immunotoxin A5-PE40 specifically binds to PSMA-positive prostate cancer cells and induces a 50% reduction of viability (IC50) at a concentration of 20 pM, while PSMA-negative cells remain unaffected. Due to its high and specific toxicity this recombinant immunotoxin is a promising candidate for therapeutic applications in patients with prostate cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer of the prostate is the most commonly diagnosed malignant tumor among males in industrialized countries and represents their second leading cause of cancer death [12]. No curative treatment currently exists for this tumor after progression beyond resectable boundaries. In most of these cases, cancer progresses despite initial treatment with androgen ablation therapy [3]. Managing advanced prostate cancer is difficult and the use of radio- and chemotherapy remains investigational. Due to the significant mortality and morbidity rate associated with the progression of this disease, there is an urgent need for new and targeted treatments.
Prostate specific membrane antigen (PSMA) is an excellent target for prostate cancer therapy because it is (a) primarily expressed in the prostate, (b) abundantly expressed as protein at all stages and even upregulated in androgen-insensitive or metastatic disease, (c) presented at the cell surface but not released into the circulation, and (d) associated with enzymatic or signaling activity [8, 13]. Besides its expression in the prostate, PSMA is also found in the neovasculature of most other solid tumors, and therefore, may serve as target for specific anti-angiogenetic drug delivery [2, 24].
PSMA is a type II membrane glycoprotein of 100 kD, with a short intracellular segment (aa 1–18), a transmembrane domain (aa 19–43) and an extensive extracellular domain (aa 44–750) [11]. As PSMA is an attractive target for imaging and immunotherapy great efforts have been undertaken to develop monoclonal antibodies (mAbs) against this antigen [16, 17, 25]. However, whereas antigen-binding mAbs could easily be found, only very few mAbs binding to cell-adherent PSMA were isolated [16]. The reason for this may be that PSMA is expressed on epithelial prostate cells as a homodimer with a specific three-dimensional structure [4]. Therefore, prostate cancer cells can only be efficiently targeted by antibodies directed against the native cell-adherent antigen [23] and only those antibodies will augment internalization, which is a precondition for delivery of intracellularly acting cytotoxic agents. To our knowledge only first generation chemically linked immunotoxins with anti-PSMA mAbs were generated [6], while no scFv against PSMA and hence also no second generation recombinant immunotoxins against this antigen have been published yet.
The toxin used for the construction of an immunotoxin in our approach was Pseudomonas exotoxin A, precisely the truncated version PE40 that lacks domain Ia, which is the binding domain for a cell surface receptor present on most mammalian cells [10]. PE40 is not toxic as long as it remains in the extracellular space. However, once linked to a scFv directed against a cell surface antigen capable of internalizing, it becomes a potent immunotoxin.
Here we describe the first fully recombinant immunotoxin against prostate cancer cells, designed with PE40 and a scFv directed against cell-adherent native PSMA. This immunotoxin is a promising candidate for further clinical evaluation in patients with prostate cancer.
Materials and methods
Cell lines
The human prostate carcinoma cell line LNCaP.FGC was used as PSMA-positive target. As PSMA-negative controls the prostate cancer cell lines DU 145 and PC-3 and the non-prostatic lines HeLa, MCF7, HCT15, MB453, K562, SW20 and Jurkat were used. For PSMA transfection, the transient packaging cell line BOSC 23 was taken. These lines and the hybridoma 7E11-C5.3 (IgG1-k, PSMA) were purchased from the American Type Culture Collection (Rockville, MD, USA). The hybridoma 3/A12 (IgG1) was obtained in our group from a Balb/c-mouse immunized with LNCaP lysate containing native PSMA (unpublished data). All cells were cultured in RPMI 1640 medium supplemented with penicillin (100 U/ml), streptomycin (100 mg/l) and 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2.
Preparation of purified PSMA
For preparing purified PSMA, about 108 LNCaP cells were washed with PBS and then lysed in PBS containing 1% NP-40 for 20 min at room temperature. After centrifugation at 10,000 g the supernatant was put on an immunoaffinity column loaded with mAb from the hybridoma 7E11-C5.3. After washing with lysis buffer PSMA was eluted with 100 mM glycine buffer (pH 2.5) containing 1% Triton X-100 and dialyzed with PBS.
Transfection of full-length PSMA into BOSC 23 cells
Full-length PSMA was cloned in two fragments (fragment 1 from 262 bp to the unique EcoRI at 1,573 bp and fragment 2 from 1,574 to 2,512 bp) into the vector pCR3.1 (Invitrogen, Karlsruhe, Germany). Transient transfection of the full-length PSMA was done with Superfect (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. After 48 h incubation the transient transfected cells were used for flow cytometric testing.
Preparation of a scFv phage display VHVL library
From cells of the hybridoma 3/A12 (IgG1, anti-PSMA) mRNA was isolated with silicagel-based membranes (Rneasy, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. From the cDNA the V region genes were amplified by PCR. For each chain 25 separate reactions were carried out by combining 25 different constant region forward primers with one corresponding reverse primer [20]. The amplified products for the light chains and the heavy chains were ligated into the phagemid pSEX (a gift from Professor M. Little, DKFZ, Heidelberg) in two cloning steps using NcoI/HindIII for the heavy chain DNA and MluI/NotI for the light chain DNA according to published methods [7].
Selection of phage particles
Panning of the phage display library was performed alternatively on PSMA-expressing LNCaP cells and on purified PSMA. For this purpose a repertoire of transformed bacteria was rescued with helper phage M13KO7 as described [19]. About 1011 recombinant phages were incubated in 2% non-fat milk/PBS with 106 LNCaP cells for 1 h at room temperature on a shaker or added to eight wells of a Maxi-Sorb microtiter plate (Nunc, Wiesbaden, Germany) which were coated with 100 μl/well of a PSMA-solution (12 μg/ml in PBS) for 2 h. After ten washing rounds with 2% non-fat milk/PBS and five rounds with PBS the bound phages were eluted with 50 mM HCl for 10 min. Eluted material was immediately neutralized by adding 1 M Tris–HCl (pH 7.5) and remaining cell debris was spun down. Phage containing supernatant was mixed with 2 ml 2xYT-medium and used to transfect 5 ml logarithmically growing E. coli TG1 cells for 30 min at 37°C before plating them on 2xYT-AG-agar-medium.
After six panning rounds, individual colonies were isolated, phage particles were rescued with helper phage M13KO7 and tested for binding to purified PSMA and LNCaP cells.
From positive clones, the V region genes were transferred into the bacterial expression vector pHOG21, which contains the sequences for c-myc- and His-tag for detection and purification of the protein [14].
Construction of the scFv-PE40 fusion protein
The sequence encoding PE40 (252–613 bp) was amplified by PCR from the plasmid pSW200 (a gift from Professor W. Wels, Frankfurt, Germany). The DNA was then ligated into the vector pHOG21 in a C-terminal position to the scFv using the restriction site XbaI. All cloning steps were performed according to standard methods in E. coli XL1-blue and the products were confirmed by sequencing.
Expression and purification of scFv and fusion protein
E. coli XL1-blue (Stratagene, La Jolla, CA, USA) cells transformed with pHOG21 plasmids were grown overnight in 2xYT-AG-medium, then diluted 1:20 and grown as 600 ml cultures at 37°C. When cultures reached OD 0.8, bacteria were pelleted by centrifugation at 1,500 g for 10 min and resuspended in the same volume of fresh 2xYT-medium containing 50 μg/ml ampicillin, 0.4 M sucrose and 1 mM IPTG. Then the bacteria were incubated at room temperature for 18–20 h. Cells were harvested by centrifugation at 5,000 g for 10 min and 4°C. To isolate soluble periplasmic proteins, the pelleted bacteria were resuspended in 30 ml of ice-cold 50 mM Tris–HCl, 20% sucrose, 1 mM EDTA (pH 8.0). After incubation for 1 h on ice, the spheroblasts were centrifuged at 20,000 g for 30 min at 4°C yielding soluble periplasmic extract in the supernatant, which was dialysed against 50 mM Tris–HCl, 1 M NaCl, (pH 7.0).
Purification was achieved by immobilized metal affinity chromatography (IMAC). This was performed using a 1 ml column of chelating Sepharose (Amersham Biosciences) charged with Cu2+ and equilibrated with a buffer containing 50 mM Tris–HCl and 1 M NaCl (pH 7.0). The periplasmatic extract was loaded on the column, washed with twenty column volumes of equilibration buffer containing 30 mM imidazole and then eluted with the same buffer containing 250 mM imidazole. Eluted material was dialyzed against PBS. Determination of the protein content was performed with the Micro BCA Protein Reagent Kit (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instructions.
Flow cytometry
Target cells were freshly harvested from tissue culture flasks and a single cell suspension was prepared in PBS with 3% FCS and 0.1% NaN3. Approximately 105 cells were incubated with 50 μl of either rescued phagemids, scFv or fusion protein for 1 h on ice. After three rounds of washing with PBS secondary antibody was added (anti-M13 mAb, Amersham Biosciences, Freiburg, Germany or anti-c-myc mAb, Roche Diagnostics, Mannheim, Germany) at 10 μg/ml and incubated for 40 min on ice. After three washings with PBS the cells were incubated with 25 μl of goat anti-mouse Ig-RPE (Becton Dickinson, Mountain View, CA, USA) for 40 min on ice. The cells were then washed repeatedly and resuspended in 200 μl of PBS containing 1 μg/ml propidium iodide, 3% FCS and 0.1% NaN3. The relative fluorescence of stained cells was measured using a FACScan® flow cytometer and the CellQuest® software (Becton Dickinson). Mean fluorescence intensity values (MFI) were considered after subtraction of the background staining with PE-labeled goat anti-mouse Ig alone.
SDS-PAGE and Western blot analysis
SDS-PAGE and Western blots were performed by standard procedures according to the manufacturer’s instructions (Invitrogen, Karlsruhe, Germany). Purified PSMA was subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes. The blots were blocked overnight in PBS containing 5% non-fat milk and incubated with scFv or scFv-PE40 at concentrations of 10 μg/ml for 1 h. Then the blots were washed five times with PBS-Tween (0.5%), incubated with a peroxidase-coupled anti-c-myc antibody (Roche Diagnostics, Mannheim, Germany) for 1 h, and then developed by using 3,3′-diaminobenzidine as substrate.
Measurement of cytotoxicity of the immunotoxin
The metabolism of the red tetrazolium salt WST to a water soluble formazan dye was determined according to the manufacturer’s instructions (Roche Diagnostics, Mannheim, Germany). Target cells were seeded at 1.5×104 cells/well of a 96-well plate and grown for 24 h until a confluent cell layer was formed. Dilutions of the recombinant immunotoxin in aliquots of 50 μl/well were added and the plates were incubated for 6, 12, 24 or 48 h at 37°C and 5% CO2. After these time points the cultures were pulsed with 15 μl/well WST reagent and incubated for 90 min at 37°C and 5% CO2. Then the spectrophotometrical absorbances of the samples were measured at 450 nm (reference 690 nm). The immunotoxin concentration required to achieve a 50% reduction in cell viability relative to that of untreated control cells (50% inhibitory concentration = IC50) was calculated.
Results
Generation of the scFv A5 against cell-adherent PSMA
For the generation of a scFv binding to cell-adherent PSMA a VHVL library was constructed from the hybridoma 3/A12 which had been obtained from a Balb/c mouse immunized with native PSMA (unpublished data). After panning of the phage display library alternatively on PSMA-expressing LNCaP cells and purified PSMA one specific positive clone, called A5 was obtained.
Construction and expression of the A5-PE40 immunotoxin
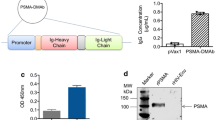
For construction of the recombinant immunotoxin, the coding sequence of PE40 from 252 to 613 bp was transferred from the vector pSW200 into the vector pHOG21 at the C-terminus of the scFv (Fig. 1). The fusion protein of about 70 kD size was purified by IMAC and about 100 μg of purified protein were routinely gained from 1 l of bacterial culture. The protein apparently retained its native folding after periplasmatic expression in bacteria, because it was not necessary to denature and renature it to obtain efficient binding and toxicity.

Construction scheme for the recombinant immunotoxin A5-PE40. His 6 , hexa-histidine-tag; c-myc, c-myc-tag; V H and V L , variable regions of the light and heavy chains of the scFv A5; pel B, leader sequence for periplasmatic protein expression; PE40, truncated ETA fragment consisting of domains II, Ib and III (aa 253–613) but lacking the receptor binding domain Ia
Comparison of the binding characteristics of the scFv A5 and the immunotoxin A5-PE40
The binding properties were determined by treating LNCaP cells with increasing concentrations of the scFv A5 and the immunotoxin A5-PE40 followed by a mouse-anti-c-myc antibody and goat anti-mouse Ig-RPE in the next step. From the resulting charts obtained by flow cytometry the A5 concentration reaching 50% saturation of PSMA sites could be estimated at 38 nM. At saturating concentrations (≥120 nM) MFI values of about 30 were measured (Fig. 2b). For A5-PE40 the concentration reaching 50% saturation of PSMA sites was determined at 40 nM. At saturating concentrations A5-PE40 reacted with viable LNCaP cells reaching MFI values of approximately 20 (Fig. 2d).

Binding of A5 and A5-PE40 to PSMA-negative DU 145 cells (a, c) and PSMA-positive LNCaP cells (b, d). Cells were stained with A5 or A5-PE40, mouse-anti-c-myc mAb, and goat anti-mouse Ig-RPE. Histograms represent logarithms of PE-fluorescence on flow cytometer. Negative control with secondary antibody alone
To evaluate the specificity of A5, PSMA-negative target cells (DU 145, PC-3, HeLa, MCF7, HCT15, MB453, K562, SW20 and Jurkat) were also analyzed, and in fact, A5 and A5-PE40 failed to bind to these cells (Fig. 2a, c shows this for DU 145 cells).
Binding of A5 and A5-PE40 to PSMA-transfected BOSC 23 cells showed concentration-dependent saturation curves (Fig. 3a, b). The MFI values were lower compared to LNCaP cells, indicating less antigen sites on the transfectants.

Binding of A5 (a) and A5-PE40 (b) to PSMA-negative BOSC 23 cells (open circle) and PSMA-transfected BOSC 23 cells (closed circle) at different concentrations. Mean fluorescence intensity values (MFI) were considered after subtraction of the background staining with secondary antibody alone
Binding of A5 and A5-PE40 to purified PSMA was tested by Western blotting. For this purpose purified PSMA was subjected to SDS-PAGE under reducing conditions and blotted. After development of the blot which was successively incubated with A5 or A5-PE40 and peroxidase labeled anti-c-myc mAb, a band at 100 kD was shown, corresponding to the monomeric PSMA (Fig. 4).

Western blot of A5 and A5-PE40 on PSMA: purified PSMA was subjected to SDS-PAGE under reducing conditions and blotted. The blot was incubated with A5 (line 1) or A5-PE40 (line 2), and peroxidase-coupled anti-c-myc mAb, and then developed with 3,3′diamino-benzidine. Line M contains a pre-stained molecular weight marker
Cytotoxicity of the immunotoxin A5-PE40 in vitro
To determine the ability of A5-PE40 to specifically inhibit the growth of PSMA-positive tumor cells, WST tests were performed. The immunotoxin was added to the cultures in a single dose, and cultures were analyzed for viability at different times (6, 12, 24 and 48 h) after addition. The immunotoxin promoted death of LNCaP cells in a time-dependent manner; highest cytotoxic effects could be observed after 48 h incubation (Fig. 5). At this time IC50 values of about 20 pM were determined. Additionally, cytotoxicity of A5-PE40 was tested on the PSMA-negative cell lines DU 145, PC-3, MCF7 and HCT 15. These cell lines only had a minimal unspecific cytotoxic background at the highest concentration of 25,000 pM (Fig. 6).

Cytotoxic effects of A5-PE40 after incubation with LNCaP cells for 6, 12, 24 and 48 h. A5-PE40 concentrations were 49 pM (black), 391 pM (grey) and 3,125 pM (white)

Effects of treatment of PSMA-positive LNCaP cells and PSMA-negative lines DU 145, PC-3, MCF7 and HCT 15. Cells were incubated for 48 h with A5-PE40 at concentrations ranging from 6 to 25,000 pM. Cell growth was measured in WST assays as described. The results are expressed as the percentage of untreated cells. Data represent mean values of three independent determinations each carried out in triplicates. SD are indicated by the error bars
The cytotoxic effect of A5-PE40 against LNCaP cells could be abrogated by an excess (300 nM) of purified parental 3/A12 antibody as shown in Fig. 7.

Inhibition of the cytotoxic effects of A5-PE40 against LNCaP cells by an excess of purified parental 3/A12 antibody. Cells were incubated with A5-PE40 at different concentrations without (black) and with addition of 3A/12 at 300 nM (grey)
Discussion
By virtue of its abundant and restricted surface expression on epithelial prostate cells, PSMA constitutes an attractive target for active and passive immunotherapies against prostate cancer. In this paper, we report a fully recombinant immunotoxin that is derived from the scFv A5 against native cell-adherent PSMA. The major findings emerging from the present study are (a) the immunotoxin shows potent and specific toxicity against PSMA-positive prostate cancer cells and (b) since PE40 is active as intracellular toxin, it can be deduced that the monovalent binding scFv A5 is capable of inducing internalization of PSMA.
Although recombinant antibody fragments and also immunotoxins against a variety of malignant cells have been reported [18], no scFv against cell-adherent PSMA and hence also no functional recombinant immunotoxin has been published yet. It has been shown by Schülke et al. [23] that PSMA is expressed on the cell surface as a homodimer with a specific three-dimensional conformation. These authors found a strong difference in the humoral immunity elicited by denatured monomeric and native dimeric forms of PSMA in as far as only the dimer efficiently elicited antibodies that recognized PSMA-expressing tumor cells.
According to these findings, we developed a mAb and successively a scFv from a mouse immunized with a lysate of LNCaP cells containing native dimeric PSMA. Additionally, the phage repertoire was selected on viable LNCaP cells expressing native PSMA.
Whereas the unconjugated scFv A5 was not cytotoxic, it acquired potent cytotoxic effects, when linked to the truncated form of Pseudomonas exotoxin A. The immunotoxin A5-PE40 was effective in the picomolar range, thus ranking among other recombinant PE40 immunotoxins [5, 15, 22]. The very low cytotoxic background on non-PSMA-expressing cells can be traced back to residual bacterial proteins or other toxic agents in the immunotoxin-preparation, because the same background could be observed in equally high concentrations with the scFv A5 alone. The cytotoxic potency of A5-PE40 against PSMA-expressing cells is remarkable and greater than the binding would predict. Other recombinant immunotoxins with scFv and Pseudomonas exotoxin A mostly showed higher binding levels on their target cells, however, IC50 values were comparable with ours [15, 22]. The reason for this could be a very high internalization rate via PSMA. It also indicates, that cross-linking of the target antigen does not seem to be necessary for the immunotoxin to be internalized.
To our knowledge there are a few approaches for targeting prostate cancer via anti-PSMA mAbs but not scFv [1, 21]. Two publications are dealing with chemically linked immunotoxins against PSMA: Fracasso et al. constructed immunotoxins by cross-linking of three anti-PSMA mAbs (J591, PEQ226.5 and PM2P079.1) to the ricin A-chain. These immunotoxins showed effects in the nanomolar range against PSMA-positive cells, whereas PSMA-negative cells were 62- to 277-fold less sensitive, thus ranking low with respect to sensitivity and specificity in comparison to our recombinant A5-PE40 [6]. Additionally, Huang et al. constructed a chemically linked immunotoxin composed of the anti-PSMA mAb E6 and deglycosylated ricin A-chain. The IC50 value on LNCaP cells was 0.06 nM, however, no data are given about the unspecific cytotoxicity [9]. The general problems of these conventional immunotoxins are not only the high immunogenicity, liver toxicity and vascular leak syndrome, but also the difficulties in producing large quantities for clinical trials. These problems might, at least in part, be overcome by recombinant immunotoxins like our A5-PE40, which are generally less immunogenic, show better tumor penetrations because of their smaller size, and permit large-scale production of pure substances. Since mouse-derived scFv also may have a rest immunogenicity, probably humanization of the framework could become necessary.
In summary, the newly constructed recombinant anti-PSMA immunotoxin A5-PE40 is both highly specific and effective against PSMA-expressing prostate cancer cells. Further evaluation will show the potential of A5-PE40 as a therapeutic agent for prostate cancer.
References
Bander NH, Trabulsi EJ, Kostakoglu L, Yao D, Vallabhajosula S, Smith-Jones P, Joyce MA, Milowsky M, Nanus DM, Goldsmith SJ (2003) Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J Urol 170:1717–1721
Chang SS, Reuter VE, Heston WD, Gaudin PB (2001) Comparison of anti-prostate-specific membrane antigen antibodies and other immunomarkers in metastatic prostate carcinoma. Urology 57:1179–1183
Chiarodo A (1991) National Cancer Institute roundtable on prostate cancer: future research directions. Cancer Res 51:2498–2505
Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ (2005) Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc Natl Acad Sci USA 102:5981–5986
Di Paolo C, Willuda J, Kubetzko S, Lauffer I, Tschudi D, Waibel R, Pluckthun A, Stahel RA, Zangemeister-Wittke U (2003) A recombinant immunotoxin derived from a humanized epithelial cell adhesion molecule-specific single-chain antibody fragment has potent and selective antitumor activity. Clin Cancer Res 9:2837–2848
Fracasso G, Bellisola G, Cingarlini S, Castelletti D, Prayer-Galetti T, Pagano F, Tridente G, Colombatti M (2002) Anti-tumor effects of toxins targeted to the prostate specific membrane antigen. Prostate 53:9–23
Fuchs P, Dubel S, Breitling F, Braunagel M, Klewinghaus I, Little M (1992) Recombinant human monoclonal antibodies. Basic principles of the immune system transferred to E. coli. Cell Biophys 21:81–91
Ghosh A, Heston WD (2004) Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J Cell Biochem 91:528–539
Huang X, Bennett M, Thorpe PE (2004) Anti-tumor effects and lack of side effects in mice of an immunotoxin directed against human and mouse prostate-specific membrane antigen. Prostate 61:1–11
Hwang J, Fitzgerald DJ, Adhya S, Pastan I (1987) Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell 48:129–136
Israeli RS, Powell CT, Corr JG, Fair WR, Heston WD (1994) Expression of the prostate-specific membrane antigen. Cancer Res 54:1807–1811
Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ (2004) Cancer statistics, 2004. CA Cancer J Clin 54:8–29
Kawakami M, Nakayama J (1997) Enhanced expression of prostate-specific membrane antigen gene in prostate cancer as revealed by in situ hybridization. Cancer Res 57:2321–2324
Kipriyanov SM, Moldenhauer G, Little M (1997) High level production of soluble single chain antibodies in small-scale Escherichia coli cultures. J Immunol Methods 200:69–77
Klimka A, Barth S, Matthey B, Roovers RC, Lemke H, Hansen H, Arends JW, Diehl V, Hoogenboom HR, Engert A (1999) An anti-CD30 single-chain Fv selected by phage display and fused to Pseudomonas exotoxin A (Ki-4(scFv)-ETA′) is a potent immunotoxin against a Hodgkin-derived cell line. Br J Cancer 80:1214–1222
Liu H, Moy P, Kim S, Xia Y, Rajasekaran A, Navarro V, Knudsen B, Bander NH (1997) Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res 57:3629–3634
Liu H, Rajasekaran AK, Moy P, Xia Y, Kim S, Navarro V, Rahmati R, Bander NH (1998) Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res 58:4055–4060
MacDonald GC, Glover N (2005) Effective tumor targeting: strategies for the delivery of armed antibodies. Curr Opin Drug Discov Devel 8:177–183
Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G (1991) By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol 222:581–597
Orum H, Andersen PS, Oster A, Johansen LK, Riise E, Bjornvad M, Svendsen I, Engberg J (1993) Efficient method for constructing comprehensive murine Fab antibody libraries displayed on phage. Nucleic Acids Res 21:4491–4498
Patri AK, Myc A, Beals J, Thomas TP, Bander NH, Baker JR Jr (2004) Synthesis and in vitro testing of J591 antibody-dendrimer conjugates for targeted prostate cancer therapy. Bioconjug Chem 15:1174–1181
Peipp M, Kupers H, Saul D, Schlierf B, Greil J, Zunino SJ, Gramatzki M, Fey GH (2002) A recombinant CD7-specific single-chain immunotoxin is a potent inducer of apoptosis in acute leukemic T cells. Cancer Res 62:2848–2855
Schulke N, Varlamova OA, Donovan GP, Ma D, Gardner JP, Morrissey DM, Arrigale RR, Zhan C, Chodera AJ, Surowitz KG, Maddon PJ, Heston WD, Olson WC (2003) The homodimer of prostate-specific membrane antigen is a functional target for cancer therapy. Proc Natl Acad Sci USA 100:12590–12595
Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C (1997) Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 3:81–85
Wang S, Diamond DL, Hass GM, Sokoloff R, Vessella RL (2001) Identification of prostate specific membrane antigen (PSMA) as the target of monoclonal antibody 107-1A4 by proteinchip; array, surface-enhanced laser desorption/ionization (SELDI) technology. Int J Cancer 92:871–876
Acknowledgments
We thank Professor W. Wels for providing the PE40 cDNA and Professor M. Little for providing the phagemid pSEX and the vector pHOG21.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wolf, P., Gierschner, D., Bühler, P. et al. A recombinant PSMA-specific single-chain immunotoxin has potent and selective toxicity against prostate cancer cells. Cancer Immunol Immunother 55, 1367–1373 (2006). https://doi.org/10.1007/s00262-006-0131-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-006-0131-0