
Abstract
Tumor immune escape variants can be identified in human and experimental tumors. A variety of different strategies are used by tumor cells to avoid recognition by different immune effector mechanisms. Among these escape routes, alteration of MHC class I cell surface expression is one of the mechanisms most widely used by tumor cells. In this review we focus our attention on the T-cell immune selection of MHC class I–deficient tumor variants. Different altered MHC class I phenotypes that originate from multiple molecular mechanisms can be identified in human tumors. MHC-deficient tumor clones can escape T-cell immune responses, but are in theory more susceptible to NK-cell–mediated lysis. In this context, we also review the controversial issue of the aberrant expression of nonclassical HLA class I molecules, particularly HLA-G, in tumors. This expression may be relevant in tumor cells that have lost the capacity to interact with NK inhibitory receptors—namely, those tumor cells with no HLA-B or HLA-C expression. Most published studies have not analyzed these possibilities and do not provide information about the complete HLA-A, HLA-B, or HLA-C molecule profiles of the tumors studied. In contrast, HLA-E has been reported to be expressed in some tumor cell lines with very low HLA-A, HLA-B, and HLA-C expression, suggesting that HLA-E may indeed, in some cases, play a role by inhibiting NK lysis of cells that otherwise would be destroyed by NK cells. Finally, we provide evidence that the status of the immune system in the tumor-bearing animal is capable of defining the MHC profile of the tumor cells. In other words, MHC class I–negative metastatic colonies are produced in immunocompetent animals, and MHC class I–positive colonies in T-cell immunodeficient individuals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tumor-associated transplantation antigens (TATAs) were defined many years ago in mice on the basis of the capacity of a particular immunocompetent host to reject a tumor transplant [47]. The assays clearly demonstrated that the immune system is capable of rejecting and destroying a tumor mass composed sometimes of many millions of tumor cells. Later, the discovery of the antigen-processing machinery (APM) used by normal and tumor cells to present peptides to T cells [61], and the identification of a variety of antigens recognized by T lymphocytes, revived interest in the role of the immune system in controlling the growth of a tumor in a particular host [5]. Tumor cells display antigen-derived peptides which, when conjugated with MHC class I molecules, are specifically recognized by the effector cells of the host immune system. Cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells are important elements in this process, and the molecular mechanisms involved in the recognition of tumor cells by cytotoxic T lymphocytes and NK cells have been partially elucidated in recent years [40].
At the beginning of primary tumor expansion, the cells are noninvasive and nonmetastatic. However, during tumor progression, mutations can lead to changes in or loss of different genes. At this point, new tumor clones appear with the capacity to invade and metastasize to distant sites. Only a small cell population in the primary tumor is now considered highly metastatic, due to genetic alterations present in each cancer cell. Metastatic cells thus acquire particular genetic, phenotypic, and biological characteristics that are not present in the primary tumor [16]. The presence of multiple genetic alterations in cancer cells indicates that these alterations accumulate during tumor progression and are responsible for differences between tumors in their particular behavior [65].
There is clear evidence that tumors frequently express antigens that are recognized by the host immune system [63]. This has been shown in experimental murine tumor models as well as in human tumors. In the latter, these antigens have been defined on the molecular level by techniques and methods for the analysis of humoral and cellular immune reactions to autologous tumor cells. A classification and listing of the tumor antigens recognized by T cells is available in different reports [49, 63].
T-cell immunoselection and tumor escape: the generation of MHC class I–deficient tumor variants
Despite an active and apparently normal immune response by a healthy immune system, tumor cells can grow, invade, and metastasize in the host [44]. Recent progress in our understanding of the interactions of tumor cells with the host immune system has led to the conclusion that tumor cells develop multiple ways to evade specific T-cell immune responses [50]. At least two major pathways have been described by which tumor cells directly escape T-cell recognition. Firstly, tumor cells can use multiple mechanisms to partially or totally down-regulate the expression of MHC class I antigens. A variety of altered HLA phenotypes have been defined in human tumors, including HLA total loss, HLA haplotype loss, HLA-specific locus down-regulation, HLA allelic losses, and a combination of these phenotypes [20, 22]. The loss of MHC class I antigens is one of the escape mechanisms found most frequently in experimental and spontaneous tumors (Fig. 1). This is not surprising, since MHC genes control the synthesis of molecules that are in many ways the center of the immune function mediated by T lymphocytes and NK cells. An increasing proportion of tumors have been found to show such alterations [21]. Total or selective losses of HLA class I antigens have been reported in different human tumor samples [20, 22].

The primary tumor is composed originally of HLA-positive cells. During tumor development HLA-negative cells appear and are immunoselected by T-lymphocyte antitumor immune responses. b The metastatic nodes are composed of highly selected tumor clones with identical or sometimes different HLA class I deficiencies
The concept that MHC class I–negative tumors are immunoselected in a host with a normal immune system, and that this selection leads to the elimination of highly immunogenic MHC class I–positive tumor cell variants by CTLs, seems difficult to prove. In fact, there is no direct evidence of the loss of HLA antigens in vivo as a consequence of antitumor effector mechanisms. However, findings in many experimental murine tumors appear to indicate that the immune system may modulate the phenotype of tumor cells (see below). In humans, the molecular mechanisms reportedly involved in loss of HLA antigens do not appear to be circumstantial findings, but may act in coordination with oncogenic factors. The total loss of HLA class I expression frequently involves two different mutations that affect both copies of the β2m gene. It is reasonable to think that the same mechanism that inactives tumor suppressor genes may also affect genes involved in the immune response to inhibit specific immune recognition. In fact, tumors with the MM phenotype select specific mutations related with cell-cycle regulations; however, at the same time these tumors are frequently highly immunogenic and show prominent lymphoid infiltration. For this reason, inactivation by mutation of the β2m gene, which is necessary for HLA class I expression, has profound effects on peptide presentation, the immediate consequence being an advantage for tumor progression in an immunocompetent host. In some cases, individual HLA class I alleles are lost, and this alteration is represented in the whole population of tumor cells. It is probable that this specific alteration leads to the inefficient presentation of immunodominant antigens.
Differential HLA class I antigen expression has been observed in successive metastases from patients. In these patients the loss of expression was due to down-regulation of the expression of the relevant HLA class I genes [34]. In other cases, tumor cells can also be immunoselected by generating tumor-specific antigen loss variants that are not presented to CD8+ cytotoxic T cells [29]. This has been shown in two metastatic cell lines from a patient with melanoma, in whom a significant difference in Melan-A/MART-1 expression was observed after immunotherapy [38]. These alterations may lead to the preferential immunoselection of melanoma antigen-negative variants during disease progression, providing a mechanism of escape from immune intervention [34].
The most frequent HLA class I alteration involves the loss of an HLA haplotype and at the same time the loss of the activatory NK receptor MICA and MICB alleles. These alterations may also favor the growth of tumor cells by simultaneous escape from T- and NK-cell recognition. Finally, down-regulation of HLA class I antigens may also be the result of a defect in the APM (TAP and LMP), which is frequently reversible by IFN-γ treatment. These APM genes are regulated by this cytokine, and the alterations may thus be the consequence of inhibition of proinflammatory cytokines that are mediated by oncogenic factors [44].
The role of nonclassical HLA class I molecules HLA-G and HLA-E in tumor escape
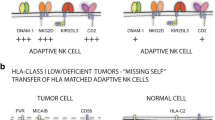
There are reports from different laboratories that HLA-G is expressed aberrantly in some human tumors tissue and cell lines [8, 45]. These findings may indicate that HLA-G expression is another mechanism of tumor escape from NK-cell attack. Such a mechanism may parallel the manner in which HLA-G protects against NK rejection of fetal tissues in the placenta [33]. However, this is still a controversial issue, since other laboratories (including ours) have not found HLA-G expression at the tumor cell surface with flow cytometry techniques [48]. However, we have found in many normal and tumoral tissues that HLA-G transcription was not followed by cell surface expression. These findings indicate strong posttranscriptional control of the expression of this gene in its different isoforms (from HLA-G1 to HLA-G5). It is also important to take into account that if a tumor cell is already expressing classical HLA-B or HLA-C molecules, NK cells are already effectively inhibited because of the strong inhibitory capacity of these two HLA locus products through their interaction with NK inhibitory receptors [40]. In these tumors, aberrant HLA-G expression is not required for tumor escape. Aberrant expression may play a role in tumor escape in some tumors, as reported in renal cell carcinomas [8], but it is far from clear whether this is so in many other tumor types such as melanomas, where HLA-G is not expressed at the cell surface [14, 43, 48].
Similarly, other nonclassical HLA class I molecules such as HLA-E might also contribute to NK tumor cell escape. It is well documented that HLA-E is widely transcribed in most tissues, and that this molecule is expressed when it combines with the leader peptides of other HLA class I molecules including HLA-G [7]. HLA-E may be used by NK cells to sense the level of expression of HLA-A, HLA-B, and HLA-C in the course of interaction with the CD94/NKG2A inhibitory receptor. This property can also be used to inhibit NK-cell–mediated tumor lysis [35]. We have found that HLA-E is expressed in some tumor cell lines with a peculiar HLA class I phenotype, e.g., in the absence of HLA-B and HLA-C expression [36] (Table 1). Selective down-regulation of HLA class I heavy chain and simultaneous HLA-E up-regulation is also seen after human cytomegalovirus (HCMV) infection, resulting in protection of target cells from NK-cell lysis [60].
Experimental evidence for T-cell immune selection in murine tumor models
The concept of immune surveillance [9, 12, 59] against cancer has been revisited in recent years. Experimental evidence in mice suggested that congenital or acquired immunosuppression was frequently associated with malignancy [37]. A good example is the spontaneous disseminated lymphoma in beige mice (the homolog of the human disorder Chediak-Higashi syndrome), in which a defect in granule formation affects multiple cell lineages (myeloid cells, cytotoxic T cells, and NK cells) [27]. It was proposed that any of these alterations might contribute to the increased incidence of tumor development. In addition, malignancy following immune suppression was often associated with Epstein-Barr virus (EBV), hepatitis B virus (HBV), human papillomavirus (HPV), or other viral infections [55, 39]. Immunosuppressed transplant patients also develop tumors of different origin, such as carcinomas and sarcomas, which can be detected by careful examination of the clinical history ([1, 6, 10, 41, 42, 64], and Gerhard Opelzt, personal communication).
Although early observations in immunodeficient mice compromised the validity of the immunosurveillance hypothesis [57], subsequent studies using better-characterized immunocompromised animals have demonstrated that the immune system may play a role in detecting the appearance of transformed cells (Table 2). Indeed, the genetic background of mice has been shown in many cases to be an important factor that determines tumor type and incidence [31]. Our laboratory recently obtained direct evidence that a particular tumor can produce MHC class I–negative or MHC class I–positive metastatic colonies depending on the immune status of the host. These findings indicate that a T-cell immune mechanism is responsible for the selection of tumor cells with a specific MHC class I phenotype [17]. Thus, the findings described below are the first indication that changes in MHC class I profile during metastatic colonization are not random, but can be reproduced in different syngenic animals (Fig. 2). These studies were performed with an H-2 class I–negative fibrosarcoma tumor clone [2, 19, 46] that generated H-2 class I–negative spontaneous lung metastases in immunocompetent BALB/c mice. In contrast, the same tumor clone produced MHC class I–positive metastatic nodes in athymic nu/nu mice [16]. This phenomenon was observed in metastatic nodules generated after a period of in vivo growth, but not in the primary tumors growing locally in the footpad. An analysis of the molecular mechanisms implicated in the origin of these MHC class I deficient metastatic nodes in immunocompetent mice suggested the coordinated suppression of multiple components of the MHC class I APM [17]. Such deficiencies are not present in metastases from immunodeficient nu/nu BALB/c mice. It has also been also reported that chemically induced sarcomas produced in nude and SCID mice were more immunogenic than similar sarcoma cells induced in congenic immunocompetent mice [13, 54, 58]. Tumors derived from nude mice were transplanted and rejected at a significantly frequent rate in normal mice [17], indicating that interaction of tumor cells with an intact immune system leads to a progressive decrease in the immunogenicity of tumor cells. CD8+ T cells were found to be necessary for this rejection, leading to the conclusion that cytotoxic T cells perform immune selection in normal mice, eliminating immunogenic tumor cell variants in the incipient tumor. Boesen et al. [4] showed that a methylcholanthrene (MCA)–induced sarcoma growing in a T-cell–immunocompetent host will eliminate highly immunogenic tumor cells that are susceptible to CD8+ T-cell–mediated lysis. In this context, it has been shown that the inoculation of immunocompetent C57BL/6 mice with mixtures of TAP1-positive and TAP1-negative cells produced tumors composed exclusively of TAP1-negative cells, indicating selection and evasion of immune surveillance by cells with the TAP deficiency [30]. These findings support the hypothesis that the MHC phenotype of tumors is influenced by the T-cell repertoire of the host, since in the absence of this T-cell pressure, the tumors “recovered” not only H-2 class I expression but also the APM functioning necessary to produce stable MHC class I molecules on the cell surface [17].

The MHC class I phenotype of a metastatic tumor clone is dependent on the immune status of the host: metastatic nodes are MHC class I–negative in immunocompetent mice, and MHC class I–positive in T-cell–immunodeficient animals
The development of gene-targeting technology has provided new evidence that lymphocytes, IFN-γ, and perforin (pfp) play a central role in providing an immunocompetent host with a mechanism of tumor surveillance against carcinogen-induced sarcomas and spontaneous tumors, a finding that also supports a role for the immune system in immunoediting tumors with low immunogenicity (tumor escape variants) [11, 32, 52]. Increased cancer susceptibility has been observed in pfp-deficient mice [53]. Perforin-deficient mice were shown to be at least 1,000-fold more susceptible to lymphomas of different lymphoid cell lineage compared with immunocompetent mice in which tumor rejection was controlled by CD8+ T lymphocytes. Approximately 50% of the pfp-deficient mice succumbed to disseminated lymphomas. In addition, an increased rate of rejection was observed in MCA-induced sarcomas produced in mice specifically deficient in Vα14 NKT cells when these tumors were transplanted into wild-type mice [53].
It was also recently shown that lymphocytes and IFN-γ play a central role in providing an immunocompetent host with a mechanism of tumor surveillance against carcinogen-induced sarcomas and spontaneous tumors [52]. For these studies, a strain of mice that completely lacked functional lymphocytes was used. Lymphocyte depletion was accomplished by inactivating a lymphocyte-specific gene called RAG2. When the mice were injected with the chemical carcinogen MCA, more than 50% developed tumors, compared with only 19% of the wild-type animals. Similar results were obtained in mice that lacked either the receptor for IFN-γ or one of the proteins required for IFN-receptor function (Stat1) [32].
The studies reviewed above provide new evidence that supports the original concept of cancer immunosurveillance. Evidence to date also supports the notion that immunoselection can confer predominance of a particular tumor escape mechanism over others. The clinically relevant mechanism in a particular patient needs to be carefully analyzed in order to develop an appropriate immunotherapeutic vaccination protocol.
Conclusions
The somatic evolution of cancer cells probably takes advantage of the generation of multiple clones that provides diversity and heterogeneity in the primary tumor lesion. The result is survival and proliferation of variants that exhibit genetic and epigenetic characteristics advantageous for growth and immune evasion. This variability sets the stage for the possible selection of immune escape variants resistant to T lymphocytes and NK-cell cytotoxicity, a process especially evident after treatment of cancer patients with increasingly effective immunotherapies. The strength and quality of the immune response determines the escape phenotype. There are clear indications that these resistant tumor variants are associated in many patients, with structural and functional alterations of human leukocyte antigen (HLA) class I molecules involved in antigen processing and presentation. These alterations can result in evasion of cytotoxic T lymphocyte (CTL) killing. In addition to down-regulation of HLA class Ia expression, the up-regulation of nonclassical HLA class Ib molecules such as HLA-E in some T-cell–resistant tumors may also favor NK immune escape.
MHC class I alterations can be defined in a wide variety of human solid tumors and cell lines. There is also recent evidence in mouse tumor models that tumor immune surveillance is not just a theory, but that immunosuppressed individuals are more susceptible to tumors. In these mice the tumor profiles, including MHC, are modulated according to the immune status of the host. We are now learning how to define these tumor escape phenotypes, including the very frequent HLA-altered profiles, and how to obtain the information needed to design tailor-made therapeutic strategies. Understanding the mechanisms and behavior of “a Darwinian natural selection” occurring during tumor progression may provide crucial information for the rational development of more efficient T-cell–directed immunotherapy.
References
Ahmed I, Hamacher KL (2002) Angiosarcoma in a chronically immunosuppressed renal transplant recipient: report of a case and review of the literature. Am J Dermatopathol 24:330
Algarra I, Gaforio JJ, Garrido A, Mialdea MJ, Pérez M, Garrido F (1991) Heterogeneity of MHC-class I antigens in clones of methylcholanthrene induced tumors: implications for local growth and metastasis. Int J Cancer 6:73
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285:727
Boesen M, Svane IM, Engel M, Rygaard J, Thomsen AR, Werdelin O (2000) CD8+ T cells are crucial for the ability of congenic normal mice to reject highly immunogenic sarcomas induced in nude mice with 3-methylcholanthrene. Clin Exp Immunol 121:1365
Boon T, Cerottini JC, Van den Eynde B, van der Bruggen P, Van Pel A (1994) Tumor antigens recognized by T lymphocytes. Annu Rev Immunol 12:337
Botti C, Seregni E, Ferrari L, Martinetti A, Bombardieri E (1998) Immunosuppressive factors: role in cancer development and progression. Int J Biol Markers 13:51
Braud V, Jones EY, McMichael A (1997) The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol 27:1164
Bukur J, Seliger B (2003) The role of HLA G for protection of human renal cell-carcinoma cells from immune-mediated lysis: implications for immunotherapies. Sem Cancer Biol 13:353
Burnet FM (1970) The concept of immunological surveillance. Prog Exp Tumor Res 13:1
Cadranel J, Naccache J, Wislez M, Mayaud C (1999) Pulmonary malignancies in the immunocompromised patient. Respiration 66:289
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991
Ehrlich P (1909) Ueber den jetzigen Stand der Karzinomforschung (About the current state of the art of cancer research). NedTijdschr Geneesk 5:273
Engel AM, Svane IM, Rigaard J, Werdelin O (1997) MCA sarcomas induced in scid mice are more immunogenic than MCA sarcomas induced in congenic, immunocompetent mice. Scand J Immunol 45:463
Frumento G, Franchello S, Palmisano GL, Nicotra MR, Giacomini P, Loke YW, Geraghty DE, Maio M, Manzo C, Natali PG, Ferrara GB (2000) Melanomas and melanoma cell lines do not express HLA-G, and the expression cannot be induced by gamma-IFN treatment. Tissue Antigens 56:30
Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH, Craft J, Yin Z (2003) Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J Exp Med 198:433
Garcia-Lora A, Algarra I, Gaforio JJ, Ruiz-Cabello F, Garrido F (2001) Immunoselection by T lymphocytes generates repeated MHC class I deficient metastatic tumor variants. Int J Cancer 91:109
Garcia-Lora A, Martinez M, Algarra I, Gaforio JJ, Garrido F (2003) MHC class I-deficient metastatic tumor variants immunoselected by T lymphocytes originate from the coordinated downregulation of APM components. Int J Cancer 106:521
Garcia-Lora A, Algarra I, Garrido F (2003) MHC class I antigens, immune surveillance and tumor immune escape. J Cell Physiol 195:346
Garrido A, Pérez M, Delgado C, Garrido ML, Rojano J, Algarra I, Garrido F (1986) Influence of class I H-2 gene expression on local tumor growth. Exp Clin Immunogenet 13:98
Garrido F, Algarra I (2001) MHC antigens and tumor escape from immune surveillance. Adv Cancer Res 83:117
Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL (1993) Natural history of HLA expression during tumour development. Immunol Today 14:491
Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar JJ, Lopez-Botet M, Duggan-Keen M, Stern P (1997) Implications for immune surveillance of altered HLA class I phenotypes in human tumors. Immunol Today 18:89
Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, Hobby P, Sutton B, Tigelaar RE, Hayday AC (2001) Regulation of cutaneous malignancy by gammadelta T cells. Science 294:605
Girardi M, Glusac E, Filler RB, Roberts SJ, Propperova I, Lewis J, Tigelaar RE, Hayday AC (2003) The distinct contributions of murine T cell receptor (TCR)gammadelta+ and TCRalphabeta+ T cells to different stages of chemically induced skin cancer. J Exp Med 198:747
Groh V, Steinle A, Bauer S, Spies T (1998) Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science 279:1737
Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T (2001) Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol 2:255
Haliotis T, Ball JK, Dexter D, Roder JC (1985) Spontaneous and induced primary oncogenesis in natural killer (NK)-cell deficient beige mutant mice. Int J Cancer 35:505
Hayashi T, Faustman DL(2002) Development of spontaneous uterine tumors in low molecular mass polypeptide-2 knockout mice. Cancer Res 62:24
Jager E, Ringhoffer M, Karbach J, Arand M, Oesch F, Knuth (1996) A Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic-T-cell responses: evidence for immunoselection of antigen-loss variants in vivo. Int J Cancer 66:470
Johnsen A, Templeton DJ, Sy MS, Harding CV (1999) Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J Immunol 163:4224
Kagi DB, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H (1994) Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369:31
Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD (1998) Demonstration of an interferon gamma dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A 95:7556
Le Bouteiller P (1997) HLA-G: on the track of immunological functions. Eur J Immunogenet 24:397
Lehmann F, Marchand M, Hainaut P, Pouillart P, Sastre X, Ikeda H, Boon T, Coulie PG (1995) Differences in the antigens recognized by cytolytic T cells on two successive metastases of a melanoma patient are consistent with immune selection. Eur J Immunol 25:340
Llano M, Lee N, Navarro F, Garcia P, Albar JP, Geraghty DE, Lopez-Botet M (1998) HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G-derived nonamer. Eur J Immunol 28:2854
Marin R, Ruiz-Cabello F, Pedrinaci S, Mendez R, Jimenez P, Geraghty DE, Garrido F (2003) Analysis of HLA-E expression in human tumors. Immunogenetics 54:767
McClain KL (1997) Immunodeficiency states and related malignancies. Cancer Treat Rev 92:39
Mendez R, Serrano A, Jager E, Maleno I, Ruiz Cabello F, Knuth A, Garrido F (2001) Analysis of HLA class I expression in different metastases from two melanoma patients undergoing peptide immunotherapy. Tissue Antigens 57:508
Meyer T, Arndt R, Nindl I, Ulrich C, Christophers E, Stockfleth E (2003) Association of human papillomavirus infections with cutaneous tumors in immunosuppressed patients. Transpl Int 16:146
Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, Moretta L (1996) Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol 14:619
Nemes B, Zalatnai A, Podder H, Jaray J, Sotonyi P Jr, Schaff Z, Foldes K, Perner F (2000) Papillary microcarcinoma of the thyroid gland in renal transplant patients. Pathol Oncol Res 6:72
Otley CC, Pittelkow MR (2000) Skin cancer in liver transplant recipients. Liver Transpl 6:253
Pangault C, Amiot L, Caulet-Maugendre S, Brasseur F, Burtin F, Guilloux V, Drenou B, Fauchet R, Onno M (1999) HLA-G protein expression is not induced during malignant transformation. Tissue Antigens. 53:335
Pardoll DM (2003) Does the immune system see tumors as foreign or self? Annual Rev Immunol 21:807
Paul P, Rouas-Freiss N, Khalil-Daher I, Moreau P, Riteau B, Le Gal FA, Avril MF, Dausset J, Guillet JG, Carosella ED (1998) HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci U S A 95:4510
Pérez M, Algarra I, Ljunggren HG, Caballero A, Mialdea MJ, Gaforio JJ, Garrido F (1990) A weakly tumorigenic phenotype with high MHC class I expression is associated with high metastatic potential after surgical removal of the primary murine fibrosarcoma. Int J Cancer 46:258
Prehn RT, Main JM (1957) Immunity to methylcholanthrene induced sarcomas. J Natl Cancer Inst 18:769
Real LM, Cabrera T, Collado A, Jimenez P, Garcia A, Ruiz-Cabello F, Garrido F (1999) Expression of HLA G in human tumors is not a frequent event. Int J Cancer 81:512
Renkvist N, Castelli C, Robbins PF, Parmiani G (2001) A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother 50:3
Restifo NP, Antony PA, Finkelstein SE, Leitner WW, Surman D, Theoret MR, Touloukian CE (2002) Assumptions of the tumor ‘escape’ hypothesis. Sem Cancer Biol 12:81
Seliger B, Cabrera T, Garrido F, Ferrone S (2002) HLA class I antigen abnormalities and immune escape by malignant cells. Sem Cancer Biol 12:3
Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Scheriber RD (2001) IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 26:1107
Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA (2000) Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med 192:755
Smyth MJ, Crowe NY, Godfrey DI (2001) NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol 13:459
Stern PL (1996) Immunity to human papillomavirus associated cervical neoplasia. Adv Cancer Res 69:175
Street SE, Cretney E, Smyth MJ (2001) Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 97:192
Stutman O (1979) Chemical carcinogenesis in nude mice: comparison between nude mice from homozygous matings and heterozygous matings and effect of age and carcinogen dose. J Natl Cancer Inst 62:353
Svane IM, Engel AM, Nielsen MB, Ljunggren HG, Rygaard J, Werdelin O (1996) Chemically induced sarcomas from nude mice are more immunogenic than similar sarcomas from congenic normal mice. Eur J Immunol 26:1844
Thomas L (1959) Discussion of cellular and humoral aspects of the hypersensitivity states. In: Lawrence HS (ed) Cellular and humoral aspects of the hypersensitive states. Hoeber-Harper, New York, p 529
Tomasec P, Braud V, Rickards C, Powell M, McSharry B, Gadola S, Cerundolo V, Borysiewicz L, McMichael A, Wilkinson G (2000) Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 287:1031
Townsend AR, Rothbard J, Gotch FM, Bahadur G, Wraith D, McMichael AJ (1986) The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell 28:959
van den Broek MF, Kagi D, Zinkernagel RM, Hengartner H (1995) Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur J Immunol 25:3514
Van den Eynde BJ, Van der Bruggen P (1997) T cell defined tumor antigens. Curr Opin Immunol 9:684
Winter P, Schoeneich G, Miersch WD, Klehr HU (1997) Tumour induction as a consequence of immunosuppression after renal transplantation. Int Urol Nephrol 29:701
Yokota J (2000) Tumour progression and metastasis. Carcinogenesis 21:497
Acknowledgements
This work was supported in part by the Fondo de Investigaciones Sanitarias, Plan Nacional de Investigacion, through Project no. BSA 2001/3080, and by the Plan Andaluz de Investigacion. We thank K. Shashok for improving the English in the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article forms part of the Symposium in Writing “Tumor escape from the immune response,” published in Vol. 53.
Rights and permissions
About this article
Cite this article
Algarra, I., García-Lora, A., Cabrera, T. et al. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: implications for tumor immune escape. Cancer Immunol Immunother 53, 904–910 (2004). https://doi.org/10.1007/s00262-004-0517-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-004-0517-9