
Abstract
There is substantial need for molecularly defined tumor antigens to prime cytotoxic T cells in vivo for cancer immunotherapy, especially in the case of tumor entities for which only a few tumor antigens have been defined so far. In this review, we present the “Tübingen approach” to identify, select, and validate large numbers of MHC/HLA class I–associated peptides derived from tumor-associated antigens. Step 1 is the identification of naturally presented HLA-associated peptides directly from primary tumor cells. Step 2 is selection of tumor-associated peptides from step 1 by differential gene expression analysis and data mining. Step 3 is validation of selected candidates by monitoring in vivo T-cell responses in the context of patient-individualized immunizations. Our approach combines methods from genomics, proteomics, bioinformatics, and T-cell immunology. The aim is to develop effective immunotherapeutics consisting of multiple tumor-associated epitopes in order to induce a broad and specific immune response against cancer cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
More than a decade has passed from initial understanding of the role of T-cell–mediated cancer immunity to the cloning of the first defined human antigen recognized by cytotoxic T cells [59]. It has since taken another decade to gather a relatively large number of clinical results with various molecularly defined vaccines and to draw informative conclusions. Despite many past and ongoing controversies, it is now widely accepted that the immune system indeed can be manipulated to specifically recognize and eliminate tumor cells as demonstrated in numerous clinical trials (reviewed by [24, 39, 45, 62]). For a long time, mainly due to a lack of known cancer antigens, active immunotherapy approaches depended completely on immunization with autologous materials including whole tumor cells, lysates, or components extracted from the patient’s tumor cells. However, these approaches are strongly limited by the restricted quantity of material available for immunization, complicated logistics, and the difficulty of defined monitoring of immune responses. The identification of tumor-associated antigens (TAAs) has opened new perspectives overcoming these limitations.
Tumor-associated antigens and peptides
Tumor cells differ from their surrounding tissue by expressing tumor-specific and tumor-associated antigens. An ideal TAA, that is a true tumor-specific antigen, is expressed only in tumor cells and not in any other tissues and is recognized by the cells of the adaptive immune system. However, such tumor-specific antigens are very rare; they usually arise from single mutations including point mutations [65], frame shift mutations [47], antisense transcripts [58], fusion proteins caused by translocation [11], or altered posttranslational modifications [52]. More commonly, TAAs are not only expressed in the tumor but are also expressed weakly in other tissues, like embryonic tissue (carcinoembryonic antigens, [57]), testes (cancer-testis antigens, [56]) or immunoprivileged sites. In some cases, they may even be expressed in tissues required for survival and well-being but to such a low extent that they are not recognized by the adaptive immune system. A different class of TAAs arises from viral infections associated with cancer—e.g., in the case of cervical cancer associated with the human papillomavirus (reviewed in [27]).
DNA microarrays have made it possible to look at the expression of thousands of genes simultaneously. The completion of the human genome project even allows analysis of the expression patterns of all known genes at the same time. This has led to the identification of a very large number of potential tumor-associated antigens. But how can this plentitude of data be leveraged for cancer immunotherapy?
Cancer immunotherapy aims at using antigens exclusively expressed or overexpressed in tumor cells, as targets for therapy. Only a few TAAs are expressed on the surface—e.g., HER-2/neu [12] or MUC-1 [19]—and may thus be targets for antibodies. Antibody-mediated therapy is also called “passive” immunotherapy because no other component of the patient’s immune system requires specific activation. So far, antibody-mediated immunity has been used very successfully for preventive vaccination against infectious diseases and was the first form of immunotherapy to enter the market for therapeutic cancer treatment—e.g., with antibodies directed to HER-2/neu found in a fraction of mammary carcinomas [30]. However, most antigens are expressed in the cytosol or organelles of the cells and thus cannot be accessed by antibodies. The immune system makes use of the cellular protein-degrading machinery and transports some of the protein fragments generated in the cytosol by the proteasome to the endoplasmic reticulum, where they are further trimmed and bound to major histocompatibility (MHC) class I molecules, called human leucocyte antigens (HLAs) in humans. HLA class I molecules are surface molecules presenting short peptides (usually 8 to 10 amino acids) derived from degraded proteins. If the peptide bound to the HLA molecule is recognized by the T-cell receptor of a cytotoxic T cell (CTL)—the HLA-associated peptide may then be called a T-cell epitope—a specific cytolytic response or a cascade of apoptotic signals as well as the secretion of various cytokines will be initiated, ultimately leading to the death of the cell presenting the peptide. However, this process can only work if the CTL was activated prior to arousal from its naïve state, a process called priming, which is assumed to be facilitated by professional antigen-presenting cells (APCs) only. Dendritic cells (reviewed in [4]) are considered the most prominent professional APCs, as they not only process antigens and present epitopes very well, but also—in their mature state—they bear high levels of costimulatory molecules at their surface thereby providing the second signal required for a naïve CD8-positive T cell to be transformed into a fully functional effector T cell. Because tumors usually cannot provide these costimulatory signals, the adaptive immune system ignores the tumor cells or, even worse, becomes tolerant toward cancer cells, falling into a state of anergy. The aim of active immunotherapy is to provide strong immunogenic tumor-associated antigens and to deliver these in a setting where effective priming of naïve T cells can be accomplished.
Application of tumor-associated peptides
HLA peptides that are derived from tumor-associated antigens—which we will refer to as tumor-associated peptides—have been shown to be very useful for priming naïve T cells to cytotoxic T cells specific for the tumor. In a clinical setting tumor-associated peptides can be loaded in vitro onto the HLA molecules of mature dendritic cells prepared from peripheral blood monocytes; e.g., by culturing these with GM-CSF, IL-4, and TNF-α; after successful pulsing, the peptide-loaded DCs are usually injected subcutaneously. Alternatively and more elegantly, peptides can be administered directly into the dermis, where the Langerhans cells, a type of dendritic cells, reside. The latter way of administration does not require the tedious preparation of DCs in vitro; instead an effective adjuvant, which enhances the immunogenic effect of the peptides, is needed.
Peptide-based immunization has many advantages over other modes of antigen delivery (e.g., proteins, viral vectors, or DNA vaccination): (1) Peptides are produced easily and rather inexpensively in clinical grade (GMP) quality; (2) Peptides have been proven safe and easy to administer in clinical settings; (3) Not only can they be used for vaccination, they are also appropriate for monitoring of specific immune responses using various in vitro and ex vivo T-cell assays. Their major disadvantage is their restriction to specific HLA alleles. However, these restrictions can be overcome: firstly, some HLA alleles like HLA-A2 in the Caucasian population or HLA-A24 in the Southeast Asian population are expressed in around half (or even more) of individuals. Secondly, with novel technologies as described in this review for the identification of tumor-associated peptides, it is becoming easier to identify peptide-based immunotherapeutics for the less frequent HLA alleles.
Strategies for identification of tumor-associated peptides
Tumor-associated peptides have been identified so far with the help of three experimental approaches dealing with the arduous task of sequence determination on the basis of different combinations of technologies.
The three experimental approaches to HLA class I peptide identification are commonly referred to as the (1) “direct” or “cellular”, (2) “genetic”, and (3) “reverse” methodologies. The “cellular” approach is based on elution of antigenic peptides from target cells and subsequent determination of peptide sequences with the help of reverse phase HPLC fractionation and Edman degradation. The direct identification of minute amounts of peptides moved into the scope of expectation with the advent of sensitive mass spectrometrical methods [13]. This approach has been advanced incrementally to unmatched accuracy and sensitivity since it became available for peptide identification. Just recently, a series of peptides from tumor cell lines [42] and primary tumor tissue [63] were identified with the help of mass spectroscopy.
The genetic approach was developed by Thierry Boon and colleagues and led to the identification of the first shared tumor antigens and corresponding peptides (reviewed in [60]). T-cell epitopes from MAGE, BAGE, GAGE, LAGE, and NY-ESO, for example, are also referred to as cancer-testis antigens, because under normal conditions their expression is limited to immunoprivileged tissues and organs, such as testis and placental trophoblasts. The majority of epitopes from the tumor-associated antigens known today were determined based on expression cloning of libraries derived from immunogenic tumor cells.
The third approach implies the use of in silico prediction methods, for this the label “reverse immunology” was coined [10]. Based on knowledge of the allelic differences (anchors, preferred residues) of HLA class I binding properties, potential HLA-associated peptides can be predicted from full-length protein sequences. Subsequently, the peptides that get a high score can be synthesized and subjected to experimental validation (HLA class I binding assays, in vitro T-cell stimulation assays). Numerous tools for reverse approaches are presently available from both academic and corporate sources. Some of the most prominent algorithms in the field are SYFPEITHI [43], PAProC [36], TEPITOPE-2000 [5], Conservatrix, EpiMatrix [34], EIS, and others (as reviewed by, e.g., De Groot et al. [14] and Nussbaum et al. [37]).
The Tübingen approach: combining genomics, peptidomics, and T-cell immunology
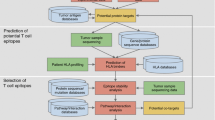
The XPRESIDENT approach developed by our group in Tübingen stands for eXpression profiling and analyis of peptide PRESentation by HLA molecules for IDEntification of New tumor antigens in combination with T-cell screening. The method is summarized in Fig. 1. It is essentially a combination of methods from genomics, HLA peptide repertoire analysis by liquid chromatography-coupled mass spectrometry (“peptidomics”), and classical as well as novel T-cell assays. This method is unique in its capability to identify a large number of HLA ligands from a specimen of frozen primary tumor material, in selection of the tumor-associated peptides from these HLA ligands based on several criteria, and validation of the selected tumor-associated peptides by in vitro T-cell assays and, where possible, in vivo, within the context of patient-individualized immunizations and monitoring of the T-cell responses. Thus, the three cornerstones of this “Tübingen approach” are identification, selection, and validation of tumor-associated peptides.

A strategy for identification, selection, and validation of tumor-associated HLA peptides for cancer immunotherapy. The approach can be used for patient-individualized treatments as well as to compose sets of “universal” tumor-associated peptides
Identification of naturally processed HLA peptides
The pure “reverse immununology” approach delivers a number of predicted peptides from any given protein sequence based on data from HLA-allele–specific motifs. Prediction is fast but not accurate: usually far more peptides than naturally processed are predicted to be presented. Even if this number can be downsized by taking preferred proteasomal cleavage sites into account (without knowing whether tumor cells express the constitutive form of the proteasome or the immunoproteasome), time-consuming experimental verification (i.e., peptide synthesis and HLA binding assays in flow cytometry) has to be carried out. This verification only reveals whether the peptide candidate has the ability to be presented by an HLA molecule; it does not answer the question whether it is naturally processed in the tumor cell. Both questions can be addressed simultaneously, if peptides are eluted directly from affinity-purified HLA molecules from primary tumor tissue. These peptides are processed and presented in the natural—i.e., patient context. However, the amount of tissue for identification of tumor-associated peptides is restricted and thus requires highly sensitive mass spectrometrical methods in combination with capillary high-performance liquid chromatography. In this way, we have reproducibly managed to identify over 100 HLA class I–associated peptides per tumor sample (minimum 5 g). The sensitivity of our analytical system is in the femtomole range, which is what enables us to obtain so large a number of peptides from individual tumor samples.
Selection of tumor-associated peptides
To obtain a selection of tumor-associated peptides which qualify as potent targets for cancer immunotherapy, we perform a differential analysis of the neoplastic, and the surrounding healthy, tissue at the molecular level. The amount of starting material for the first line of analysis ranges from whole organs, such as a kidney (in the case of renal cell carcinoma) to small biopsy samples. In the case of organ resection, both malignant and normal material is available for analysis in sufficient amounts. In the case of tissue biopsy, amounts are limited, and it can be difficult to obtain material from normal tissues. Moreover, accurate analysis requires that there be no mix-up of the sample material of interest with unrelated healthy cell types from directly adjacent tissue, which sometimes can only be obtained after enrichment procedures. Enrichment can be achieved by several methods: e.g., laser capture microdissection, cell sorting by magnetic beads, or fluorescence-assisted cell sorting. Expression analysis of single tumor cells by DNA array technology is possible and has been described [26]. A critical issue here is the fidelity of quantitative representation of the original individual mRNA species after PCR-based cDNA amplification.
Large-scale screening for differences between tumor and normal cells can be carried out at the level of DNA, protein, mRNA, or HLA ligands. Comparative expression profiling of a tumor and the corresponding autologous normal tissue granted by DNA microarray technology [31, 50] is an excellent method for identifying large numbers of candidate tumor-associated antigens from individual tumor samples [6]. Using DNA chip technology, genome-wide expression analysis of all genes is possible within a few hours. In addition, expression data for each gene in almost all normal human tissues and organs is available from in-house gene expression databases generated from tissue-specific mRNA pooled from a large number of donors. Coherent and reliable data sets can be generated on the basis of high-quality sample preparations and the use of DNA array systems with low margins of variation. In this way, expression of every antigen in the tumor sample can be compared with expression levels of the same antigen in the surrounding tissue and almost all other tissues. This data is especially important for the selection of the appropriate antigens suitable for immunization with respect to autoimmunity and T-cell tolerance.
A differential direct “HLA peptidome” analysis of the tumor and the corresponding normal cells would be ideal, since differences in antigen processing between tumor and normal tissues are detected in this setting. Epitopes might exist which are only presented on the tumor cells, although the source protein is present in equal amounts in both cell types. There is increasing evidence that the peptide pool generated by the immunoproteasome differs from that produced by the constitutive proteasome [55], an interesting aspect in the context of tumors exposed to local release of interferon-γ (IFN-γ). However, differential HLA peptidome analysis is difficult to achieve due to the much lower amounts of peptides obtained by HLA immunoprecipitation and elution compared with the number of genes covered by DNA arrays. We are developing a method called “QUALITEA” which allows quantitative comparative analysis of tumor and normal tissue by isotope labeling of all peptides eluted from tumor cells and mixing of these peptides with all peptides eluted from normal cells. The restriction of this method is the requirement for equal amounts of normal tissue. QUALITEA allows us to directly quantify differentially displayed peptides by mass spectrometry analysis (Lemmel et al., in press).
Apart from the considerations mentioned above, such as overexpression in tumor and limited expression in other tissues as determined by genome- or peptidome-based analysis, further criteria can be considered for the selection of tumor-associated peptides: known role in carcinogenesis, or reported immunogenicity of the gene product. The analysis of existing databases provides additional help. Serological identification of antigens by recombinant expression cloning (SEREX) [48] provides a list of TAAs recognized by antibodies. Some antigens identified by this method were shown to be recognized concomitantly by CD8+ T cells [22]. Although there is no direct link between detection of antibodies against a particular TAA and a CD8+ T cell recognizing the same TAA, it could at least be stated that antigens identified by SEREX are in general immunogenic. Further useful and immediately available information regarding tissue distribution of a gene, virtual Northern blots, genomic hybridization data, gene annotations, etc. is available from various public databases [2, 17, 18, 28, 41, 61, 67].
Another different form of “selection” is inherent in the method of identification itself. Even if more than 100 different HLA peptides are identified from one tumor sample, this represents less than 10% of the HLA peptidome of the tumor cell because several thousand different sequences per cell can be expected to be presented [54]. However, not all presented peptides will be able to elicit a successful signal to the specific T cell because most peptides are presented in very low copy numbers. The most well-defined parameters for TCR engagement are ligand density and TCR affinity. The TCR must be engaged by the corresponding HLA-peptide complex long enough to elicit a complete set of signaling events required for efficient T-cell activation [7]. Even for high-affinity ligands, it has been shown that an HLA-peptide–complex density below the activation threshold does not lead to successful T-cell activation. It is safe to assume that due to the sensitivity limit of our analytical setup, those HLA peptides that are abundantly presented by the tumor cells and thus, have higher chances to elicit a T-cell response in vivo because of their high ligand density, are the first to be found. This is especially valid if peptides derived from self-proteins are used in immunization approaches; as for these self-peptides, that might also be present in low copies on normal tissues, a high ligand density is required.
An alternative way—where selection precedes identification—is the use of prediction algorithms in combination with identification of predicted epitopes directly from primary tumor tissue. We have named this method “Predict, Calibrate, and Detect” [51]. Epitopes are predicted from a sequence of a known tumor-associated protein using the algorithms SYFPEITHI [43] and PAProC [36]. In a second step, predicted candidate peptides are synthesized to be used for calibration of the capillary liquid chromatography–mass spectrometrical system. In a third step, these peptides are then searched for among the HLA-associated peptides eluted from primary tumor material as described above. In this way, the existence of every predicted epitope is verified or negated.
Validation of tumor-associated CTL epitopes
Naturally processed and presented HLA peptides are not always CTL epitopes. Only the successful elicitation of a human CTL response in vitro or, better, in vivo delivers the final validation for a tumor-associated HLA peptide.
Antigen-specific CTLs can be induced in vitro by synthesizing the identified tumor-associated peptides and pulsing these on mature dendritic cells generated in vitro. These peptide-pulsed DCs are then cocultured with fresh PBMCs from healthy donors and restimulated with the peptide, thereby promoting in vitro priming of naïve precursor CD8+ T cells. Peptide-specific activity of CTLs generated in this way can be measured by various T-cell assays: e.g., standard 51Cr-release assay to determine the capability of in vitro primed T cells to kill peptide-pulsed target cells in a peptide-specific manner and additionally, tumor cell lines—without additional peptide-pulsing—expressing the corresponding antigen from which the HLA peptide had been derived.
The superior way to validate the quality of tumor-associated peptides as an immunotherapeutic is to monitor peptide-specific CD8+ T-cell responses in immunized patients. The ultimate challenge then is to correlate the outcome of the clinical results with these immunological responses.
In the cases where peptides have been injected subcutaneously or intradermally, alone or in combination with adjuvant, measure of delayed-type hypersensitivity (DTH) reaction at the site of injection can be performed. This is the only in vivo test available to demonstrate the induction of cellular immunity. Several clinical trials have shown a correlation between intensity of DTH reaction and the expansion of peptide-specific CD8+ T cells in the blood of immunized patients [15]. Analysis of the T-cell infiltrate present at the DTH site can indicate which cells have been recruited by the immunization [23]. There are several ex vivo methods available to estimate CTL activity and frequency prior to and after vaccination.
The most common technique to quantify the activity of CTLs ex vivo is the IFN-γ ELISpot assay [35]. It is based on the detection of IFN-γ secretion by antigen-stimulated T cells. The secreted cytokine is detected by a specific antibody coated on a nitrocellulose surface and is then visualized as a spot using a second enzyme-linked anti–IFN-γ antibody. This assay has been used in numerous clinical trials and does not strictly require prior in vitro expansion of the T cells. Thus, besides delivering functional measure of T-cell stimulation, this assay also allows an estimation of the frequency of antigen-specific CD8+ T cells. If purified CD8+ T cells are used, or if an in vitro presensitization step with the peptide is added, the sensitivity of the method can reach 1–10 IFN-γ–secreting cells in 50,000 [8, 23]. Alternatively, the production of IFN-γ and other cytokines in single T cells in vitro can also be measured after permeabilization of cells and subsequent intracellular staining with cytokine-specific antibodies [9]. Used in multiple stainings in flow cytometry (FACS), this technique determines which subpopulation (CD4+, CD8+, CD45RA/RO, etc.) is actually responding to the antigenic stimulus. The cytokine release assay which detects secreted cytokine by cell surface–bound cytokine-specific antibodies, allows simultaneous sorting of the responding population by magnetic separation [40]. Another method for ex vivo detection of cytokine production by T cells is based on the detection of cytokine mRNA up-regulation in stimulated T cells by real-time quantitative reverse transcriptase (RT)–PCR [25]. After a short incubation of cells with the tumor-associated peptide, production of cytokine (e.g., IFN-γ) mRNA can be determined. This method provides indirect information about the specific T-cell frequency, and activation is detected at the mRNA level only. However, because it necessitates a limited number of cells, it constitutes an attractive assay for screening antigen-reactive CD8+ T cells.
The most accurate method to determine the frequency of CD8+ T cells ex vivo is using soluble fluorescent tetrameric HLA-peptide complexes commonly known as tetramers [1]. The detection limit with tetramers varies from 1 in 2,000 to 1 in 10,000 CD8+ T cells (frequency 0.05–0.01%). For most of the tumor antigen-derived peptides, this sensitivity is insufficient to identify specific T cells directly ex vivo. Nevertheless, tetramers are powerful tools to detect and enumerate specific T cells. Tetramer-positive cells can be characterized using relevant markers of T-cell subpopulations. In particular, the surface expression of CD45RA/RO, CCR7, and LFA-1 defines different effector/memory subpopulations [29, 49]. This type of phenotypical study helps to further define efficient antitumor CTLs and to optimize vaccination protocols [53]. Tetramer staining can also be coupled to intracellular or cytokine release assays (see above). This type of test indicates whether the specific T cells are also functional, as generally not all tetramer-positive cells are able to produce cytokines in vitro upon antigenic stimulation. Finally, tetramer technology allows the sorting of pure peptide-specific T-cell populations [16]. These cells can be studied further—for example, by clonotypic analysis of TCR transcripts—or expanded in vitro for functional assays.
A crucial question is where to look for specific T cells. For practical reasons, the search for specific T cells has been performed using patient blood samples. However, monitoring of the peripheral T-cell response only might underestimate the immunogenicity of a tumor-associated peptide. In particular, specific recruitment of activated T cells to the tumor sites causes these cells to be undetectable in the blood [66]. Tumor-specific T cells can be found at high frequency among tumor-infiltrating lymphocytes [38] and in tumor-draining lymph nodes [44]. Although these studies cannot be conducted on a routine basis, they should nonetheless help to evaluate the quality of the T-cell response induced by vaccination.
Clinical development of peptide-based immunotherapeutics
The first results from therapeutic vaccinations with tumor-associated peptides were published in 1995 and 1996. A peptide from MAGE-3 led to the first clinically validated response to peptide-based immunotherapy [32], showing that tumor regression was within the scope of expectation for this novel therapeutic approach. Jaeger et al. were first in reporting results from the use of multiple melanoma-associated peptides in man [21]. A variety of approaches to identify optimal routes and modes of delivery—e.g., the use of GM-CSF [20], dendritic cells [32], or peptides mixed with Incomplete Freund’s Adjuvant combined with coadministration of high-dose IL-2 [46]—went along with clinical trials using tumor-associated peptides, partly showing very encouraging clinical responses. Still, the results of the majority of clinical trials using one or two tumor-associated peptides have been disappointing. Although specific immune responses were induced in a variable proportion of immunized patients ranging up to 80% in a few reports, objective clinical responses were often not seen in more than 10–20% of patients. We assume that the lack of substantial clinical responses is mainly due to two reasons: the first is that when immunizing with one or two antigens it is usually not clear whether the antigen of choice is expressed in the tumor. For instance, it is known that the well-characterized tumor-associated antigen HER-2/neu is expressed in only approximately 20% of all mammary carcinoma patients [64]; still HER-2/neu is often recognized as a “universal” tumor antigen. In approximately 20 renal cell tumors, we ourselves have never detected expression of HER-2/neu (unpublished data). The second reason might be that a number of immune evasion mechanisms exist, allowing the tumor to evade the immune response [33, 62]: termination of T-cell activation via CTLA-4, IL-2–mediated activation-induced cell death (AICD) of T cells, CD4+CD25+ regulatory T cells suppressing the action of T cells, or release of immunosuppressive TGF-β by the tumor itself. Although these immune evasion mechanisms can be overcome by the use of antibodies and cytokines, down-regulation of single antigens or even HLA alleles can still occur due to the selective pressure of the therapeutic antigen. Therefore, it seems essential to direct the cytotoxic T-cell response toward many targets simultaneously, ideally only using peptides from antigens expressed in the tumor and presented by different HLA allelic variants.
Just recently, Banchereau and co-workers obtained insights into how the interplay between multiple tumor-associated peptides, APCs, and homing might be orchestrated with striking effectiveness. Stage IV melanoma patients were vaccinated with autologous dendritic cells prepared from CD34+ precursor cells and loaded with four well-characterized HLA-A2-restricted melanoma-associated peptides. Interestingly, there was a strong correlation between the number of peptide-specific T-cell responses and the clinical outcome in terms of tumor regression and survival rates. Ten out of 18 patients included in this study were nonprogressors and almost all of the patients in this group showed more than three different T-cell responses to the peptides vaccinated, while seven out of eight of the patients in the progressor group showed no or only up to two different T-cell responses [3] (personal communication with J. Banchereau).
Although these results are very encouraging for the whole field of cancer vaccination, there is not a sufficient number of tumor-associated peptides available to compose multipeptide vaccines for tumor indications other than melanoma, as well as for HLA alleles other than HLA-A2. Therefore, it is crucial to identify, select, and validate large numbers of tumor-associated peptides in different tumor entities and for different HLA alleles.
A first reasonable step toward the development of multipeptide cancer vaccines would be to compose a set of about six to ten tumor-associated peptides per HLA allele derived from antigens expressed in a majority of cancer patients. If these peptides were derived from tumor-associated antigens expressed in a minimum of 60% of the patients, six peptides per set should be sufficient to elicit at least three different T-cell responses in more than 80% of the patients participating in the trial (own calculation, unpublished). Such peptide sets could be designed for the most frequent HLA alleles. One step further would be the combination of several peptide sets for different HLA alleles depending on the HLA typing of the patient. Thus, a typical member of the Caucasian population might receive a combination of, e.g., an HLA-A2 and HLA-B7 set, while most East Asians would receive a combination of, e.g., HLA-A2 and HLA-A24. The ultimate challenge would be to select peptides from a large validated tumor-associated peptide library depending on the individual gene expression pattern in the tumor of the patient. In this patient-individualized setting, although logistically more complicated, the patient would receive an immunotherapeutic where every peptide was known to be derived from an antigen definitely overexpressed in the patient’s tumor. With molecularly defined human tumor antigens, this would be the first individualized cancer therapy approach “off-the-shelf.”
References
Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM (1996) Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94–96
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29
Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, Taquet S, Coquery S, Wittkowski KM, Bhardwaj N, Pineiro L, Steinman R, Fay J (2001) Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res 61:6451–6458
Banchereau J, Paczesny S, Blanco P, Bennett L, Pascual V, Fay J, Palucka AK (2003) Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Ann N Y Acad Sci 987:180–187
Bian H, Reidhaar-Olson JF, Hammer J (2003) The use of bioinformatics for identifying class II-restricted T-cell epitopes. Methods 29:299–309
Boer JM, Huber WK, Sultmann H, Wilmer F, von Heydebreck A, Haas S, Korn B, Gunawan B, Vente A, Fuzesi L, Vingron M, Poustka A (2001) Identification and classification of differentially expressed genes in renal cell carcinoma by expression profiling on a global human 31,500-element cDNA array. Genome Res 11:1861–1870
Boniface JJ, Davis MM (1995) T-cell recognition of antigen. A process controlled by transient intermolecular interactions. Ann N Y Acad Sci 766:62–69
Britten CM, Meyer RG, Kreer T, Drexler I, Wolfel T, Herr W (2002) The use of HLA-A*0201-transfected K562 as standard antigen-presenting cells for CD8(+) T lymphocytes in IFN-gamma ELISPOT assays. J Immunol Methods 259:95–110
Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W (2000) Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood 96:3102–3108
Celis E, Tsai V, Crimi C, DeMars R, Wentworth PA, Chesnut RW, Grey HM, Sette A, Serra HM (1994) Induction of anti-tumor cytotoxic T lymphocytes in normal humans using primary cultures and synthetic peptide epitopes. Proc Natl Acad Sci U S A 91:2105–2109
Clark RE, Dodi IA, Hill SC, Lill JR, Aubert G, Macintyre AR, Rojas J, Bourdon A, Bonner PL, Wang L, Christmas SE, Travers PJ, Creaser CS, Rees RC, Madrigal JA (2001) Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR-ABL b3a2 fusion protein. Blood 98:2887–2893
Coussens L, Yang-Feng TL, Liao YC, Chen E, Gray A, McGrath J, Seeburg PH, Libermann TA, Schlessinger J, Francke U (1985) Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 230:1132–1139
Cox AL, Skipper J, Chen Y, Henderson RA, Darrow TL, Shabanowitz J, Engelhard VH, Hunt DF, Slingluff CL Jr (1994) Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science 264:716–719
De Groot AS, Sbai H, Aubin CS, McMurry J, Martin W (2002) Immuno-informatics: Mining genomes for vaccine components. Immunol Cell Biol 80:255–269
Disis ML, Schiffman K, Gooley TA, McNeel DG, Rinn K, Knutson KL (2000) Delayed-type hypersensitivity response is a predictor of peripheral blood T-cell immunity after HER-2/neu peptide immunization. Clin Cancer Res 6:1347–1350
Dunbar PR, Ogg GS, Chen J, Rust N, van der BP, Cerundolo V (1998) Direct isolation, phenotyping and cloning of low-frequency antigen-specific cytotoxic T lymphocytes from peripheral blood. Curr Biol 8:413–416
Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210
Haverty PM, Weng Z, Best NL, Auerbach KR, Hsiao LL, Jensen RV, Gullans SR (2002) HugeIndex: a database with visualization tools for high-density oligonucleotide array data from normal human tissues. Nucleic Acids Res 30:214–217
Hayes DF, Mesa-Tejada R, Papsidero LD, Croghan GA, Korzun AH, Norton L, Wood W, Strauchen JA, Grimes M, Weiss RB (1991) Prediction of prognosis in primary breast cancer by detection of a high molecular weight mucin-like antigen using monoclonal antibodies DF3, F36/22, and CU18:a Cancer and Leukemia Group B study. J Clin Oncol 9:1113–1123
Jaeger E (1996) Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. Int J Cancer 67:54–62
Jaeger E, Bernhard H, Romero P, Ringhoffer M, Arand M, Karbach J, Ilsemann C, Hagedorn M, Knuth A (1996) Generation of cytotoxic T-cell responses with synthetic melanoma-associated peptides in vivo: implications for tumor vaccines with melanoma-associated antigens. Int J Cancer 66:162–169
Jager E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jager D, Arand M, Wada H, Noguchi Y, Stockert E, Old LJ, Knuth A (1998) Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1:definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med 187:265–270
Jager E, Maeurer M, Hohn H, Karbach J, Jager D, Zidianakis Z, Bakhshandeh-Bath A, Orth J, Neukirch C, Necker A, Reichert TE, Knuth A (2000) Clonal expansion of Melan A-specific cytotoxic T lymphocytes in a melanoma patient responding to continued immunization with melanoma-associated peptides. Int J Cancer 86:538–547
Jager E, Jager D, Knuth A (2002) Clinical cancer vaccine trials. Curr Opin Immunol 14:178–182
Kammula US, Lee KH, Riker AI, Wang E, Ohnmacht GA, Rosenberg SA, Marincola FM (1999) Functional analysis of antigen-specific T lymphocytes by serial measurement of gene expression in peripheral blood mononuclear cells and tumor specimens. J Immunol 163:6867–6875
Klein CA, Seidl S, Petat-Dutter K, Offner S, Geigl JB, Schmidt-Kittler O, Wendler N, Passlick B, Huber RM, Schlimok G, Baeuerle PA, Riethmuller G (2002) Combined transcriptome and genome analysis of single micrometastatic cells. Nat Biotechnol 20:387–392
Konya J, Dillner J (2001) Immunity to oncogenic human papillomaviruses. Adv Cancer Res 82:205–238
Lash AE, Tolstoshev CM, Wagner L, Schuler GD, Strausberg RL, Riggins GJ, Altschul SF (2000) SAGEmap: a public gene expression resource. Genome Res 10:1051–1060
Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM (1999) Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med 5:677–685
Leyland-Jones B (2002) Trastuzumab: hopes and realities. Lancet Oncol 3:137–144
Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL (1996) Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol 14:1675–1680
Marchand M, Weynants P, Rankin E, Arienti F, Belli F, Parmiani G, Cascinelli N, Bourlond A, Vanwijck R, Humblet Y (1995) Tumor regression responses in melanoma patients treated with a peptide encoded by gene MAGE-3. Int J Cancer 63:883–885
Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S (2000) Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol 74:181–273
Martin W, Sbai H, De Groot AS (2003) Bioinformatics tools for identifying class I-restricted epitopes. Methods 29:289–298
Miyahira Y, Murata K, Rodriguez D, Rodriguez JR, Esteban M, Rodrigues MM, Zavala F (1995) Quantification of antigen specific CD8+ T cells using an ELISPOT assay. J Immunol Methods 181:45–54
Nussbaum AK, Kuttler C, Hadeler KP, Rammensee HG, Schild H (2001) PAProC: a prediction algorithm for proteasomal cleavages available on the WWW. Immunogenetics 53:87–94
Nussbaum AK, Kuttler C, Tenzer S, Schild H (2003) Using the World Wide Web for predicting CTL epitopes. Curr Opin Immunol 15:69–74
Panelli MC, Bettinotti MP, Lally K, Ohnmacht GA, Li Y, Robbins P, Riker A, Rosenberg SA, Marincola FM (2000) A tumor-infiltrating lymphocyte from a melanoma metastasis with decreased expression of melanoma differentiation antigens recognizes MAGE-12. J Immunol 164:4382–4392
Parmiani G, Castelli C, Dalerba P, Mortarini R, Rivoltini L, Marincola FM, Anichini A (2002) Cancer immunotherapy with peptide-based vaccines: what have we achieved? where are we going? J Natl Cancer Inst 94:805–818
Pittet MJ, Zippelius A, Speiser DE, Assenmacher M, Guillaume P, Valmori D, Lienard D, Lejeune F, Cerottini JC, Romero P (2001) Ex vivo IFN-gamma secretion by circulating CD8 T lymphocytes: implications of a novel approach for T cell monitoring in infectious and malignant diseases. J Immunol 166:7634–7640
Pruitt KD and Maglott DR (2001) RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res 29:137–140
Ramakrishna V, Ross MM, Petersson M, Gatlin CC, Lyons CE, Miller CL, Myers HE, McDaniel M, Karns LR, Kiessling R, Parmiani G, Flyer DC (2003) Naturally occurring peptides associated with HLA-A2 in ovarian cancer cell lines identified by mass spectrometry are targets of HLA-A2-restricted cytotoxic T cells. Int Immunol 15:751–763
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219
Romero P, Dunbar PR, Valmori D, Pittet M, Ogg GS, Rimoldi D, Chen JL, Lienard D, Cerottini JC, Cerundolo V (1998) Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J Exp Med 188:1641–1650
Rosenberg SA (2001) Progress in human tumour immunology and immunotherapy. Nature 411:380–384
Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, Wunderlich JR, Parkhurst MR, Kawakami Y, Seipp CA, Einhorn JH, White DE (1998) Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 4:321–327
Saeterdal I, Bjorheim J, Lislerud K, Gjertsen MK, Bukholm IK, Olsen OC, Nesland JM, Eriksen JA, Moller M, Lindblom A, Gaudernack G (2001) Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci U S A 98:13255–13260
Sahin U, Tureci O, Schmitt H, Cochlovius B, Johannes T, Schmits R, Stenner F, Luo G, Schobert I, Pfreundschuh M (1995) Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci U S A 92:11810–11813
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712
Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470
Schirle M, Keilholz W, Weber B, Gouttefangeas C, Dumrese T, Becker HD, Stevanovic S, Rammensee HG (2000) Identification of tumor-associated MHC class I ligands by a novel T cell-independent approach. Eur J Immunol 30:2216–2225
Skipper JC, Hendrickson RC, Gulden PH, Brichard V, Van Pel A, Chen Y, Shabanowitz J, Wolfel T, Slingluff CL Jr, Boon T, Hunt DF, Engelhard VH (1996) An HLA-A2-restricted tyrosinase antigen on melanoma cells results from posttranslational modification and suggests a novel pathway for processing of membrane proteins. J Exp Med 183:527–534
Speiser DE, Lienard D, Pittet MJ, Batard P, Rimoldi D, Guillaume P, Cerottini JC, Romero P (2002) In vivo activation of melanoma-specific CD8(+) T cells by endogenous tumor antigen and peptide vaccines: a comparison to virus-specific T cells. Eur J Immunol 32:731–741
Stevanovic S, Schild H (1999) Quantitative aspects of T cell activation--peptide generation and editing by MHC class I molecules. Semin Immunol 11:375–384
Toes RE, Nussbaum AK, Degermann S, Schirle M, Emmerich NP, Kraft M, Laplace C, Zwinderman A, Dick TP, Muller J, Schonfisch B, Schmid C, Fehling HJ, Stevanovic S, Rammensee HG, Schild H (2001) Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J Exp Med 194:1–12
Traversari C, van der BP, Luescher IF, Lurquin C, Chomez P, Van Pel A, De Plaen E, Amar-Costesec A, Boon T (1992) A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J Exp Med 176:1453–1457
Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J (1995) Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst 87:982–990
Van Den Eynde BJ, Gaugler B, Probst-Kepper M, Michaux L, Devuyst O, Lorge F, Weynants P, Boon T (1999) A new antigen recognized by cytolytic T lymphocytes on a human kidney tumor results from reverse strand transcription. J Exp Med 190:1793–1800
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den EB, Knuth A, Boon T (1991) A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254:1643–1647
van der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, Chapiro J, Van Den Eynde BJ, Brasseur F, Boon T (2002) Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev 188:51–64
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial analysis of gene expression. Science 270:484–487
Waldmann TA (2003) Immunotherapy: past, present and future. Nat Med 9:269–277
Weinschenk T, Gouttefangeas C, Schirle M, Obermayr F, Walter S, Schoor O, Kurek R, Loeser W, Bichler KH, Wernet D, Stevanovic S, Rammensee HG (2002) Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer Res 62:5818–5827
Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM (2003) Expression of the HER1–4 family of receptor tyrosine kinases in breast cancer. J Pathol 200:290–297
Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, Beach D (1995) A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269:1281–1284
Yamshchikov GV, Barnd DL, Eastham S, Galavotti H, Patterson JW, Deacon DH, Teates D, Neese P, Grosh WW, Petroni G, Engelhard VH, Slingluff CL Jr (2001) Evaluation of peptide vaccine immunogenicity in draining lymph nodes and peripheral blood of melanoma patients. Int J Cancer 92:703–711
Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, Vogelstein B, Kinzler KW (1997) Gene expression profiles in normal and cancer cells. Science 276:1268–1272
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented at the first Cancer Immunology and Immunotherapy Summer School, 8–13 September 2003, Ionian Village, Bartholomeio, Peloponnese, Greece.
Rights and permissions
About this article
Cite this article
Singh-Jasuja, H., Emmerich, N.P.N. & Rammensee, HG. The Tübingen approach: identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol Immunother 53, 187–195 (2004). https://doi.org/10.1007/s00262-003-0480-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-003-0480-x