
Abstract
Purpose
Differentiation among parkinsonian syndromes may be clinically challenging, especially at early disease stages. In this study, we used 18F-FDG-PET brain imaging combined with an automated image classification algorithm to classify parkinsonian patients as Parkinson’s disease (PD) or as an atypical parkinsonian syndrome (APS) at the time when the clinical diagnosis was still uncertain. In addition to validating the algorithm, we assessed its utility in a “real-life” clinical setting.
Methods
One hundred thirty-seven parkinsonian patients with uncertain clinical diagnosis underwent 18F-FDG-PET and were classified using an automated image-based algorithm. For 66 patients in cohort A, the algorithm-based diagnoses were compared with their final clinical diagnoses, which were the gold standard for cohort A and were made 2.2 ± 1.1 years (mean ± SD) later by a movement disorder specialist. Seventy-one patients in cohort B were diagnosed by general neurologists, not strictly following diagnostic criteria, 2.5 ± 1.6 years after imaging. The clinical diagnoses were compared with the algorithm-based ones, which were considered the gold standard for cohort B.
Results
Image-based automated classification of cohort A resulted in 86.0% sensitivity, 92.3% specificity, 97.4% positive predictive value (PPV), and 66.7% negative predictive value (NPV) for PD, and 84.6% sensitivity, 97.7% specificity, 91.7% PPV, and 95.5% NPV for APS. In cohort B, general neurologists achieved 94.7% sensitivity, 83.3% specificity, 81.8% PPV, and 95.2% NPV for PD, while 88.2%, 76.9%, 71.4%, and 90.9% for APS.
Conclusion
The image-based algorithm had a high specificity and the predictive values in classifying patients before a final clinical diagnosis was reached by a specialist. Our data suggest that it may improve the diagnostic accuracy by 10–15% in PD and 20% in APS when a movement disorder specialist is not easily available.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Clinical differentiation among parkinsonian syndromes (Parkinson’s disease (PD), multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD)) may be challenging in early disease stages. They all present with parkinsonism which is a clinical syndrome of bradykinesia in combination with rest tremor, rigidity, or both [1]. Specific signs of individual atypical parkinsonian syndromes (MSA, PSP, and CBD) may be absent initially. Although there is no disease-modifying treatment available for parkinsonian syndromes, correct diagnosis is important for various reasons. Symptomatic surgical and device-aided treatments are beneficial mostly in PD [2] and incorrect diagnosis may lead to unnecessary and potentially harmful interventions; moreover, enrolment of misdiagnosed patients in clinical trials may lead to inaccurate results. Furthermore, the prognosis in these syndromes differs [2] and correct diagnosis is important for management of the long-term patients’ care. The definitive diagnosis of parkinsonism can still be made only by pathohistological examination of the brain tissue [3]. Clinicopathological studies have shown low diagnostic accuracy in early disease with positive predictive value for PD of only 53% for symptom durations under 5 years [4]. PD is often clinically overdiagnosed and atypical parkinsonian syndromes underdiagnosed by general neurologists in early stages [5].
Presynaptic dopaminergic imaging (e.g., dopamine transporter imaging) has only limited role in differentiating among various parkinsonian syndromes, given that the presynaptic dopaminergic system is affected in atypical parkinsonian syndromes as well as classical PD [6]. The postsynaptic D2 receptor imaging may be used for differentiation among PD and atypical parkinsonian syndromes, but it is not specific enough for individual case diagnosis [7]. However, regional changes in brain metabolism vary among parkinsonisms and it has been shown that positron emission tomography with 18F-fluoro-2-deoxyglucose (18F-FDG-PET) can be used for the differentiation among them. Visual reading and statistical parametric mapping analysis of brain 18F-FDG-PET images are common supplementary diagnostic tools for diagnosis of parkinsonian syndromes and monitoring of disease progression [6, 8,9,10,11]. Additionally, network analysis of 18F-FDG-PET images using a spatial covariance technique known as scaled subprofile model (SSM) based on the principal component analysis (PCA) [12, 13] has been used to identify specific disease-related metabolic brain patterns in PD [14,15,16,17], MSA [18], PSP [18], and CBD [19].
For the differential diagnosis of parkinsonisms in clinical practice, an automated probabilistic algorithm which utilizes the expressions of multiple disease-related metabolic patterns was developed by Tang et al. [20] and validated in a subsequent study by Tripathi et al. [21]. In the first step, this algorithm differentiates among patients with PD and atypical parkinsonian syndrome (APS), while in the second one, it differentiates between MSA and PSP. High specificity (90–97%) and positive predictive values (PPV, 85–98%) of diagnostic ability of this logistic algorithm were reported in previous studies [20, 21].
In this study, we aimed to first validate the differential diagnostic accuracy of the abovementioned automated probabilistic algorithm on a new European cohort (cohort A analysis) and then to evaluate its usefulness in the “real-life” clinical settings (cohort B analysis). For the first aim, we compared the automated algorithm-based imaging diagnosis with the final clinical diagnosis made by the movement disorder specialist for PD, MSA, and PSP patients in cohort A. To fulfill the second aim, we tested the accuracy of the clinical diagnosis of parkinsonian syndromes made by general neurologists with the imaging diagnosis made by the automated probabilistic algorithm in cohort B patients. Our hypothesis was that the automated 18F-FDG-PET-based diagnosis is more specific than a general neurologist and therefore of particular value in situations when specialized movement disorder service is not available.
Methods
Subjects
Cohort A consisted of 66 consecutive patients with uncertain clinical diagnosis of parkinsonism who underwent diagnostic 18F-FDG-PET imaging between October 2010 and August 2015. At least 1 year after imaging, patients were reexamined by a movement disorder specialist blinded to patients’ history and imaging results and a final clinical diagnosis was made based on the clinical diagnostic criteria for PD (UK Brain Bank Criteria) [22], MSA [23], and PSP [24]. The PSP was diagnosed using the 1996 NINDS-SPSP criteria [24]; therefore, all the included PSP patients are very likely to be affected by PSP-Richardson syndrome (and not by any other PSP subtype) as defined by the latest MDS-PSP criteria [25]. According to the diagnostic criteria used [22, 24], patients with neuroleptic drug exposure, pronounced vascular or other structural lesions, head trauma or encephalitis history, family history of parkinsonism, long remission or unilateral involvement after 3 years as well as patients with uncertain or alternative diagnosis were excluded. In one patient, the diagnosis of PD was made by postmortem pathohistological brain examination. The average time interval between 18F-FDG-PET imaging and the final clinical diagnosis was 2.2 ± 1.1 years (mean ± SD). Only patients with final clinical or histopathological diagnosis of PD, MSA, and PSP—the three most common parkinsonian syndromes [26]—were included in the analysis.
Cohort B consisted of 71 consecutive patients with uncertain clinical diagnosis of parkinsonism who were also referred to 18F-FDG-PET by their neurologists between October 2010 and August 2015. Unlike in cohort A patients, the final clinical diagnosis of patients in cohort B was made by a general neurologist in a “real life” setting, not strictly following the diagnostic criteria but rather their clinical expertise [22,23,24]. The average time interval between 18F-FDG-PET imaging and the clinical diagnosis was 2.5 ± 1.6 years for the cohort B patients. As in cohort A, only patients with clinical diagnosis of PD, MSA, and PSP were included in the analysis.
All the patients were recruited from the Department for Neurology, University Medical Center Ljubljana.
In addition, 20 age- and gender-matched normal controls (NC), who were described before [17], underwent 18F-FDG-PET imaging and were included in the analysis for the purposes of z-transforming subject scores of the three metabolic patterns for individual patients.
The study was approved by the National Medical Ethics Committee of the Republic of Slovenia. Written informed consent was obtained from the participating subjects.
18F-FDG-PET imaging
Subjects were instructed to fast overnight prior to PET imaging. 18F-FDG-PET was performed at the Department of Nuclear Medicine at UMC Ljubljana. A total of 250 MBq of 18F-fluorodeoxyglucose (FDG) was injected intravenously; afterwards, the subjects were placed to rest in a dark room with eyes closed. Brain scans were acquired using Siemens Biograph mCT PET/CT scanner 30 min after FDG injection. Images were reconstructed to 400 × 400 × 110 matrix with voxel size 1.02 × 1.02 × 3mm3 using OSEM+PSF+TOF reconstruction algorithm and 4 mm post processing Gaussian filter. Patients’ dopaminergic medication was not withheld prior to 18F-FDG-PET.
Image processing
18F-FDG-PET images of each patient were converted to SPM Analyze format using MRIConvert software (http://lcni.uoregon.edu/~jolinda/MRIConvert/). They were spatially normalized into a standard Montreal Neurological Institute (MNI) space and smoothed using a 10 × 10 × 10 mm FWHM Gaussian filter using SPM5 software (Wellcome Trust Centre for Neuroimaging), running in MATLAB 7.0 (MathWorks Inc.).
Calculation of brain 18F-FDG-PET-based network expression values
The subjects’ expressions of the previously determined and validated characteristic metabolic patterns were calculated. The PD-related pattern (PDRP) [14], MSA-related pattern (MSARP) [18], and PSP-related pattern (PSPRP) [18] were all identified at Feinstein Institute for Medical Research, New York. These subjects’ expressions of the three networks were calculated in individual subjects using SCANVP software (http://www.feinsteinneuroscience.org at Center for Neuroscience, Feinstein Institute for Medical Research, NY, USA) as described elsewhere [12]. Expression values for each pattern were z-transformed using mean and standard deviation of subject scores from the NC group [17].
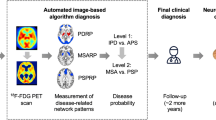
Differential diagnosis of individual parkinsonian patients by automated logistic algorithm
An automated algorithm, developed in the previous study [20], was used to diagnostically classify each of the patients from cohorts A and B, based on the PDRP, MSARP, and PSPRP expressions (i.e., subject scores) computed from the 18F-FDG-PET scan of each patient. In the first stage, patients were classified as PD, APS, or indeterminate parkinsonism. This was done based on the calculation of probability for PD or APS (cutoff values for PD and APS were 81% and 79%, as determined in the previous study [20]). In the second stage, the patients who were classified as APS in the first step underwent further classification as MSA, PSP, or indeterminate APS. Probability cutoff values of 74% and 55% were used for MSA and PSP, respectively, whereas subjects with lower probability than the cutoff probabilities of both diseases were considered as indeterminate [20].
Comparison of an automated algorithm-based diagnosis with the final clinical diagnosis
In cohort A, the algorithm-based diagnosis of individual patients was compared with the patients’ final clinical diagnosis, which was made by a movement disorder specialist at least 1 year after 18F-FDG-PET scan and blinded to the previous clinical workup. The final clinical diagnosis was considered a diagnostic gold standard in this cohort.
In cohort B, the clinical diagnosis made by a general neurologist was compared with the automated algorithm-based diagnosis, which was considered a diagnostic gold standard in this cohort.
For each cohort sensitivity, specificity, PPV, and negative predictive values (NPV) were calculated on both diagnostic stages and reported in Results. Clinical characteristics of patients with either indeterminate or incongruent diagnoses by clinical and automated imaging diagnoses were presented individually in the Electronic Supplement Materials for cohort A (Tables S1 and S2) and cohort B (Tables S3 and S4).
Automated classification of the excluded patients from cohorts A and B (i.e., patients with alternative or unclear clinical diagnosis) was performed as well and is presented in the Electronic Supplement Materials (Figs. S1 and S2).
Statistical analysis
Demographic data, disease duration at 18F-FDG-PET scan and at final clinical diagnosis, the time interval between 18F-FDG-PET scan and clinical diagnosis, as well as metabolic pattern expression values, across the disease groups in each cohort were compared using one-way ANOVA test followed by post hoc Bonferroni’s multiple comparisons tests performed in GraphPad Prism v7 for Windows (GraphPad Software, San Diego, CA, USA). To calculate disease probabilities based on metabolic pattern scores for individual patients, logistic regression analysis was performed in SAS 9.3 (SAS Institute Inc., Cary, NC, USA). For all comparisons, p value ˂ 0.05 was considered statistically significant.
Results
Cohort A
Of the 66 patients, whose final clinical diagnosis was made by a movement disorder specialist (Fig. 1a), ten were excluded from further analysis due to final clinical diagnosis other than PD, MSA, or PSP (two were diagnosed with CBD, two with dementia with Lewy bodies (DLB), one with subjective cognitive disorder, one with iatrogenic parkinsonism, and four with uncertain clinical diagnosis). Of the remaining 56 patients, 43 were clinically diagnosed with PD, 6 with MSA, and 7 with PSP. REM sleep behavior disorder was reported by 12/43 PD and 1/6 MSA patients; one PD patient reported vivid visual hallucinations. For all 56 cohort A patients, their mean age at the time of 18F-FDG-PET scan was 67.1 ± 8.5 years and mean disease duration was 4.0 ± 2.6 years. Final clinical diagnosis was made 2.2 ± 1.1 years after 18F-FDG-PET scan and 6.3 ± 2.9 years after the onset of their symptoms. No significant differences were found across the PD, MSA, and PSP groups in age at disease onset (p = 0.40; one-way ANOVA), age at 18F-FDG-PET (p = 0.13), disease duration at 18F-FDG-PET (p = 0.68), the disease duration at clinical diagnosis (p = 0.34), or in the time interval between 18F-FDG-PET and the final clinical diagnosis (p = 0.23). The demographic and clinical data of cohort A patients are summarized in Table 1.

Study design and results of automated algorithm-based classification for cohorts A and B
Metabolic network expressions in PD, MSA, and PSP groups
PDRP, MSARP, and PSPRP expression scores of individual patients in cohort A are presented in Figs. 2 a and 3 a. A significant difference in PDRP expression (subject scores) was found across parkinsonian syndromes (Fig. 2a, left; F(3, 72) = 12.6, p ˂ 0.0001, one-way ANOVA). The PDRP expression was higher in PD patients compared with NC (p = 0.0003; post hoc Bonferroni test) and MSA group (p < 0.0001); it was also higher in PSP patients compared with MSA (p = 0.002). There was no difference in PDRP expression between MSA and NC (p = 0.22) and between PD and PSP groups (p = 1.0). The difference between PSP and NC was only marginal (p = 0.06).

Metabolic brain patterns’ expressions for PDRP, MSARP, and PSPRP measured as subject scores across cohort A in which the diagnostic gold standard was clinical diagnosis) (a) and cohort B in which the diagnostic gold standard was algorithm-based diagnosis (b). Subject scores of cohort A and cohort B patients are presented. Mean values and standard errors are shown. Differences in all network subject scores across clinical diagnoses groups were statistically significant (one-way ANOVA, p ˂ 0.0001). Asterisks above graphs represent statistically significant differences among various groups (post hoc Bonferroni test). *p ˂ 0.05, **p ˂ 0.01, ***p ˂ 0.001, ****p ˂ 0.0001. Differences among groups that are not marked with asterisk are not significant

Tree-dimensional graph presenting a combination of PDRP, MSARP, and PSPRP Z-scores for PD, MSA, and PSP patients from cohort A (gold standard clinical criteria based diagnosis) and cohort B (gold standard algorithm-based diagnosis). IND, indeterminate parkinsonism
A significant difference in MSARP subject scores was present across the three groups (Fig. 2a, middle; F(3, 72) = 22.1, p ˂ 0.0001, one-way ANOVA), with higher pattern expressions for both MSA and PSP groups relative to NC or PD group (p ˂ 0.0001; post hoc Bonferroni test). There was no difference in MSARP expression between MSA and PSP groups or between PD and NC groups (p = 1.0).
We also found a significant difference in PSPRP expression across groups (Fig. 2a, right; F(3, 72) = 35.3, p ˂ 0.0001, one-way ANOVA). Subject scores in the PSP group were higher than those in all other groups (p ≤ 0.0001; post hoc Bonferroni test). Moreover, PSPRP expression in MSA patients was significantly higher than in both the NC and PD groups (p = 0.006 and p = 0.01). There was no significant difference between NC and PD patients (p = 1.0).
Comparison of an automated algorithm-based diagnosis with the final clinical diagnosis made by movement disorder specialist
Among the 56 parkinsonian patients from cohort A, an automated algorithm classified 38 patients as PD (37 clinically diagnosed as PD, one as MSA) and twelve as APS (one clinically diagnosed as PD, five as MSA, six as PSP); the remaining six patients (10.7%) were classified as indeterminate parkinsonism (five clinically diagnosed as PD, one as PSP) at the first stage. Detailed classification of all the subjects (along with the excluded ones) is presented in the Electronic Supplement Material (Fig. S1). Compared with the final clinical diagnosis which was a gold standard in cohort A, the algorithm achieved 86.0% sensitivity, 92.3% specificity, 97.4% PPV, and 66.7% NPV for PD, as well as 84.6% sensitivity, 97.7% specificity, 91.7% PPV, and 95.5% NPV for APS (Table 2).
Twelve patients classified as APS were further analyzed at the second stage; four were classified as MSA (all congruent with clinical diagnosis) and six as PSP (five congruent with clinical diagnosis, one clinically diagnosed as PD); two (16.7%) were indeterminate (one clinically diagnosed as MSA and one as PSP). The algorithm achieved 80.0% sensitivity, 100% specificity, 100% PPV, and 87.5% NPV for MSA, and 83.3% sensitivity, specificity, PPV, and NPV for PSP (Table 2).
Cohort B
Among the 71 patients in cohort B whose clinical diagnosis was made at the regular clinical visit by the general neurologist (Fig. 1b), 28 were excluded from further analysis due to clinical diagnosis other than PD, MSA, or PSP: four had clinical CBD, six had clinical DLB, one had frontotemporal dementia, one probable Creutzfeldt-Jakob disease and diagnosis was clinically unclear in 16 cases. Demographic and clinical data of the 43 patients included in the analysis of cohort B are presented in Table 1. Their mean age was 64.5 ± 10.5 years at the time of 18F-FDG-PET scan and their disease duration was 6.3 ± 4.2 years. The clinical diagnosis was made 8.7 ± 4.8 years after the onset of the symptoms and 2.5 ± 1.6 years after 18F-FDG-PET scan. No significant differences across the PD, MSA, and PSP were found in age at disease onset (p = 0.73; one-way ANOVA), at 18F-FDG-PET (p = 0.18), or at the disease duration at 18F-FDG-PET (p = 0.08). The disease duration at the clinical diagnosis differed significantly across groups (p = 0.01); it was significantly longer for PD compared with MSA (p = 0.01, post hoc Bonferroni’s test). A significant difference was found also in the time interval between 18F-FDG-PET and clinical diagnosis (p = 0.03); it was shorter in MSA patients compared with PD (p = 0.03, post hoc Bonferroni’s test).
Comparison of the routine clinical diagnosis made by a general neurologist with an automated algorithm-based diagnosis
Of the 43 cohort B patients, the automated algorithm classified 19 patients as PD (18 of those were clinically diagnosed as PD, one as PSP), 17 as APS (two were clinically diagnosed as PD, six as MSA, nine as PSP), and seven (16.3%) as indeterminate parkinsonism (two were clinically PD and five MSA) at the first stage. Detailed classification of all the subjects (along with the excluded ones) is presented in the Electronic Supplement Material (Fig. S2). Using the automated image-based diagnosis as gold standard for comparison, the clinical diagnosis of cohort B patients resulted in 94.7% sensitivity, 83.3% specificity, 81.8% PPV, and 95.2% NPV for PD, and 88.2% sensitivity, 76.9% specificity, 71.4% PPV, and 90.9% NPV for APS (Table 2).
Seventeen APS patients were further analyzed in the second stage. Among them, five were classified as MSA (four were clinically diagnosed as MSA and one as PSP) and twelve as PSP (two clinically diagnosed as PD, two as MSA, and eight as PSP). Clinical diagnosis compared with the gold standard automated classification resulted in 80.0% sensitivity, 83.3% specificity, 66.7% PPV, and 90.9% NPV for MSA, and 66.7% sensitivity, 80.0% specificity, 88.9% PPV, and 50.0% NPV for PSP (Table 2).
In addition, metabolic network expressions in cohort’s B PD, MSA, and PSP groups as categorized by the algorithm are presented in Fig. 2 b. A significant difference in PDRP, MSARP, and PSPRP expression was found across these parkinsonian syndromes (p ˂ 0.0001, one-way ANOVA).
Discussion
In this study, we tested the diagnostic utility of the specific metabolic brain patterns, which were identified previously in parkinsonian patients using 18F-FDG-PET brain imaging and the multivariate image analysis. We first re-validated the diagnostic accuracy of the automated metabolic brain pattern-based algorithm in 56 parkinsonian patients (cohort A), by comparing it with the final clinical diagnosis, which was done strictly following the diagnostic criteria. In this cohort A analysis, the clinical diagnosis was the gold standard. Second, we compared the clinicians’ routine diagnosis of parkinsonism (cohort B) with the diagnosis obtained from the automated and algorithm-based diagnostic procedure. The algorithm calculates the diagnostic probability for PD, MSA, and PSP based on the measurement of the expression of specific metabolic brain pattern characteristic for these three syndromes. In this cohort B analysis, the automated algorithm-based diagnosis was the gold standard.
The differential diagnosis algorithm [20] was thus validated in a new European cohort in the first part of the study, followed by the test of its utility in a “real-life” clinical setting in the second one.
In both cohorts, we evaluated the differences in the expressions of the three patterns (PDRP, MSARP, and PSPRP) between the groups of parkinsonian patients. We found a highly significant difference in the three network expressions when comparing their expressions among corresponding patient groups and NC (p ˂ 0.0001), which is consistent with the original identification studies [14, 18].
In the validation cohort A analysis, however, a single pattern did not differentiate among all patient groups (Fig. 2). To some extent, this may be due to the effects of dopaminergic therapy, which was not discontinued in these patients before imaging. Dopaminergic therapy may lower PDRP expression in PD patients to the levels closer to those seen in PSP [27].
To be able to discriminate among all parkinsonian syndromes based on the disease-related metabolic brain patterns, the expression levels (subject scores) for all the three patterns (i.e., PDRP, MSARP, and PSPRP) should be quantified in each individual patient (Fig. 3). The automated algorithm described previously [20] computes these measures, which are then submitted to the logistic regression for individual subject categorization. In cohort A, the automated algorithm-based analysis accurately discriminated PD patients from APS (first stage), as well as MSA from PSP (second stage). The findings agree with previously published observations from North America [20] and India [21]. The specificity for PD in cohort A was 92% (compared with North America and India, 98% and 95%, respectively), and PPV was 97% (compared with 97% and 88%, respectively). APS were also accurately categorized, with a specificity of 98% and a PPV 92% comparable with that observed in the previously published North American and Indian samples (specificity 98% and 95%, PPV 97% and 88%).
Interestingly, there were only two incongruent cases (3.6%) in our cohort A sample in which the image-based categorization differed from the clinical diagnosis (Table S1). The first case was a patient diagnosed clinically as parkinsonian-type MSA (MSA-P), whom the algorithm classified as PD. The second was clinically diagnosed as PD but classified as PSP according to the algorithm. Both incongruent patients had substantial cortical atrophy on MRI, which may have confounded image-based classification in these individuals [21]. That said, clinical diagnoses are not always upheld at autopsy. Indeed, the full impact of discrepancies such as these cannot be appreciated without autopsy confirmation.
Eight patients (14%) from cohort A were classified as indeterminate by the logistic algorithm (Table S2): six at the first and two at the second stage (both being classified correctly as APS). Interestingly, features such as disease duration at FDG-PET and at final diagnosis, as well as the time interval between FDG-PET and final diagnosis, did not differ among indeterminate vs. definitively categorized patients. Of eight indeterminate patients, three had moderate brain atrophy, whereas two had no atrophy on MRI. It is possible that in these cases, the disease process had not matured sufficiently to point to a specific diagnostic classification at the time of imaging [20]. In any event, the percentages of indeterminate cases at level 1 (11%) and level 2 (17%) in cohort A were similar to those reported previously in the earlier samples [20, 21]. As in the earlier samples, indeterminate cases at level 1 were evenly distributed clinically between PD and APS (level 1), or between MSA and PSP (level 2).
While a high degree of concordance was seen between the clinical diagnoses of the movement disorder specialist following the diagnostic criteria and the pattern-based imaging classifications, the same cannot be said for the diagnoses made by general neurologists in group B.
Previous clinicopathological studies have shown low PPV (38–65%) of early clinical diagnosis for PD (< 5 years) which later increases to 85% [4, 28]. The diagnostic accuracy is considerably higher in specialist movement disorder settings with PPV up to 99% for PD, 86% for MSA, and 80% for PSP [29]. Another clinicopathological study revealed high sensitivity (89%) but low specificity (58%) of general neurologists for PD while it was opposite for APS with high specificity (99% for MSA and 100% PSP) and low sensitivity (64% and 53%, respectively). These findings indicate that PD may be overdiagnosed while APS is clinically underdiagnosed in clinical practice [5].
Comparing the discriminative measures from cohorts A and B (keeping in mind different gold standards and the study design, which prevent us from directly paralleling the results), we may conclude that the automated algorithm was more specific while clinical neurologist was more sensitive for PD (the specificity was 92.3 by automated algorithm and 83.3% by clinical neurologists, while sensitivity was 86.0% and 94.7%, respectively). The cutoff values for automated classification were intentionally selected in favor of higher specificity (> 90%) against sensitivity (> 80%) in the original study [20]; as in the clinical settings, 18F-FDG-PET is used for confirmation rather than for screening. While the specificity and PPV of automated classification in APS were high as well, discriminative measures of “real-life” clinical diagnosis were considerably lower.
The clinicians’ high sensitivity with lower specificity for PD is consistent with pathological studies mentioned above. These results are not surprising since the PD is more prevalent than the APS, red flag signs often appear late, and the consequences of misdiagnosis are usually not very harmful since there are no disease-modifying drugs available and symptomatic dopaminergic therapy may be tried in APS, although without prolonged efficacy, as is the case in PD [2]. It is different in the subgroup of patients who are considered for advanced therapies (e.g., device-assisted and surgical treatments). The risk of complications is higher in these therapies [30] and APS patients do not benefit from them as PD patients do [31]. In this subgroup of patients, high specificity and PPV for PD are especially important and can be achieved either by specialist in movement disorder, or as suggested by our results, by the automated algorithm analysis based on metabolic brain characteristics, particularly in situations where movement disorder specialist may not be available. Although the design of our study did not allow for a direct comparison of both cohorts due to distinct gold standards, our data suggest that the algorithm may help non-expert general neurologists to improve their diagnostic accuracy by approximately 10–15% in PD patients and by 20% in APS. With considerably high PPV and specificity, the algorithm may be especially useful for patient selection in clinical trials of new therapies for PD or APS [32]. Moreover, while by design the algorithm is currently applied to only differentiate among PD, MSA, and PSP, we found that it classified all 8 DLB patients as PD (2 in cohort A and 6 in cohort B) in the additional analysis of the patients with alternative diagnoses (Figs. S1 and S2). This result suggests that the use of the image-based algorithm could potentially be extended to the differential diagnosis of DLB, although it remains a working hypothesis that requires formal testing in future studies with larger samples.
There were six (14%) incongruent cases in cohort B. Two patients classified as PSP were clinically diagnosed with MSA-P. The crucial clinical characteristic in both cases that probably steered a clinician to make the diagnosis of MSA was orthostatic hypotension that may occasionally be present also in PSP [33]. Another two patients classified as PSP were clinically diagnosed with PD. An early cognitive decline was present in the first case and poor L-dopa responsiveness in the second one; however, they did not convince the clinician against making the diagnosis of PD. One patient classified as PD was clinically diagnosed with PSP. Patient had early dementia, postural instability, and vertical gaze palsy suggesting PSP, but also REM sleep behavior disorder, which is commonly associated with synucleinopathies and is rare in PSP [34]. One patient with automated diagnosis of MSA was clinically diagnosed with PSP. In that case, the review of the documentation did not reveal any of the typical clinical features of MSA. Seven cohort B patients were classified as indeterminate at level 1: one with clinical diagnosis of cerebellar type MSA (MSA-C) and four with MSA-P. MSA-P patients exhibited substantial structural changes; in three cases, a moderate or severe cortical atrophy and in one posttraumatic atrophy was seen on brain MRI. The final two indeterminate patients were clinically diagnosed with PD.
There are some noteworthy limitations of our study. Firstly, the computerized algorithm applied to our patients’ 18F-FDG-PET images relies on disease-related metabolic patterns that were identified and validated by the technique’s originators. Therefore, differences in PET instrumentation and reconstruction algorithms among the two centers may influence pattern expression values (subject scores) for individual cases [35]. Even so, calibration of patient values against healthy control subjects scanned at the study site helped to correct for systematic differences of this sort [36]. The use of locally derived patterns for PD, MSA, and PSP [17] may help in this regard in the future. Secondly, there is an imbalance in the proportion of PD and APS patients in cohorts A and B. In cohort A, there is predominance of PD patients with the PD:APS ratio 3.3:1, which is considerably higher than in the originator’s North American cohort (1.4:1) [20]. Cohort B is more balanced and comparable with the North American cohort (PD:APS ratio 1.1:1). The imbalance, however, does not influence sensitivity and specificity, which are features of the diagnostic test and not related to the different prevalence, but it may, according to the Bayes theorem, result in artificially higher PPV and lower NPV in cohort A. Moreover, the number of APS patients is relatively small as these disorders are rare. Therefore, caution is required in interpreting the percentage of APS patients. Thirdly, the study is limited by the absence of pathological confirmation of the “gold standard” diagnoses. In the current study, only a single patient underwent autopsy, which interestingly confirmed the image-based classification (PD) but contradicted the treating neurologist’s diagnosis (MSA). Nonetheless, firm conclusions regarding the accuracy of the image-based algorithm relative to the true pathological gold standard cannot be made without systematic correlations in autopsied patients.
Most of the neuro-nuclear diagnostic centers nowadays routinely use univariate voxel-based parametric Z-score maps to differentiate among neurodegenerative disorders [11]. The algorithm presented here represents another approach to further enhance the 18F-FDG-PET-based diagnosis. In the future, we intend to compare the algorithm-based diagnosis with the one made by an experienced neuro-nuclear medicine specialist relying on voxel-based Z-score maps.
Conclusion
Based on our results and previously published studies, metabolic network-based logistic algorithm is a reliable tool to improve early diagnostic accuracy in patients with uncertain parkinsonism. We believe that it is suitable for use in clinical practice as well as an inclusion/exclusion tool for clinical trials.
While the accuracy of clinical diagnosis of parkinsonian patients is high in specialized movement disorder centers, it is considerably lower in the general practice. The 18F-FDG-PET is nowadays widely available tool and the automated diagnostic algorithm may contribute to the correct diagnosis by considerably increasing the diagnostic accuracy, for 10–15% in PD and 20% in APD, in regions where movement disorder specialist is not easily accessible.
References
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30:1591–601.
Stamelou M, Bhatia KP. A typical parkinsonism diagnosis and treatment. Neurol Clin Elsevier Inc. 2015;33:39–56.
Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. Cold Spring Harbor Laboratory Press; 2012;2.
Adler CH, Beach TG, Hentz JG, Shill HA, Caviness JN, Driver-Dunckley E, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology. 2014;83:406–12.
Joutsa J, Gardberg M, Röyttä M, Kaasinen V. Diagnostic accuracy of parkinsonism syndromes by general neurologists. Parkinsonism Relat Disord. 2014;20:840–4.
Niccolini F, Politis M. A systematic review of lessons learned from PET molecular imaging research in atypical parkinsonism. Eur J Nucl Med Mol Imaging. 2016;43:2244–54.
Hellwig S, Amtage F, Kreft A, Buchert R, Winz OH, Vach W, et al. [18F]FDG-PET is superior to [123I]IBZM-SPECT for the differential diagnosis of parkinsonism. Neurology. 2012;79:1314–22.
Tripathi M, Dhawan V, Peng S, Kushwaha S, Batla A, Jaimini A, et al. Differential diagnosis of parkinsonian syndromes using F-18 fluorodeoxyglucose positron emission tomography. Neuroradiology. 2013;55:483–92.
Juh R, Kim J, Moon D, Choe B, Suh T. Different metabolic patterns analysis of parkinsonism on the18F-FDG PET. Eur J Radiol. 2004;51:223–33.
Huang C, Tang C, Feigin A, Lesser M, Ma Y, Pourfar M, et al. Changes in network activity with the progression of Parkinson’s disease. Brain Oxford University Press. 2007;130:1834–46.
Caminiti SP, Alongi P, Majno L, Volontè MA, Cerami C, Gianolli L, et al. Evaluation of an optimized [ 18 F]fluoro-deoxy-glucose positron emission tomography voxel-wise method to early support differential diagnosis in atypical Parkinsonian disorders. Eur J Neurol. 2017;24:687–e26.
Spetsieris PG, Ma Y, Dhawan V, Eidelberg D. Differential diagnosis of parkinsonian syndromes using PCA-based functional imaging features. Neuroimage. 2009;45:1241–52.
Spetsieris PG, Eidelberg D. Scaled subprofile modeling of resting state imaging data in Parkinson’s disease: methodological issues. Neuroimage. 2011;54:2899–914.
Ma Y, Tang C, Spetsieris PG, Dhawan V, Eidelberg D. Abnormal metabolic network activity in Parkinson’s disease: test—retest reproducibility. J Cereb Blood Flow Metab. 2007;27:597–605.
Wu P, Wang J, Peng S, Ma Y, Zhang H, Guan Y, et al. Metabolic brain network in the Chinese patients with Parkinson’s disease based on 18F-FDG PET imaging. Parkinsonism Relat Disord. 2013;19:622–7.
Teune LK, Renken RJ, Mudali D, De Jong BM, Dierckx RA, Roerdink JBTM, et al. Validation of parkinsonian disease-related metabolic brain patterns. Mov Disord. 2013;28:547–51.
Tomše P, Jensterle L, Grmek M, Zaletel K, Pirtošek Z, Dhawan V, et al. Abnormal metabolic brain network associated with Parkinson’s disease: replication on a new European sample. Neuroradiology. 2017;59:507–15.
Eckert T, Tang C, Ma Y, Brown N, Lin T, Frucht S, et al. Abnormal metabolic networks in atypical parkinsonism. Mov Disord. 2008;23:727–33.
Niethammer M, Tang CC, Feigin A, Allen PJ, Heinen L, Hellwig S, et al. A disease-specific metabolic brain network associated with corticobasal degeneration. Brain. 2014;137:3036–46.
Tang CC, Poston KL, Eckert T, Feigin A, Frucht S, Gudesblatt M, et al. Differential diagnosis of parkinsonism: a metabolic imaging study using pattern analysis. Lancet Neurol. 2010;9:149–58.
Tripathi M, Tang CCC, Feigin A, De Lucia I, Nazem A, Dhawan V, et al. Automated differential diagnosis of early parkinsonism using metabolic brain networks: a validation study. J Nucl Med. 2016;57:60–6.
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4.
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6.
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9.
Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32:853–64.
Erkkinen MG, Kim M-O, Geschwind MD. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2018;10:a033118.
Ko JH, Lee CS, Eidelberg D. Metabolic network expression in parkinsonism: clinical and dopaminergic correlations. J Cereb Blood Flow Metab. 2017;37:683–93.
Beach TG, Adler CH. Importance of low diagnostic accuracy for early Parkinson’s disease. Mov Disord. 2018;33:1551–4.
Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125:861–70.
Merola A, Espay AJ, Romagnolo A, Bernardini A, Rizzi L, Rosso M, et al. Advanced therapies in Parkinson’s disease: long-term retrospective study. Parkinsonism Relat Disord. 2016;29:104–8.
Shih LC, Tarsy D. Deep brain stimulation for the treatment of atypical parkinsonism. Mov Disord. 2007;22:2149–55.
Schindlbeck KA, Eidelberg D. Network imaging biomarkers: insights and clinical applications in Parkinson’s disease. Lancet Neurol. 2018;17:629–40.
Wenning GK, Scherfler C, Granata R, Bösch S, Verny M, Chaudhuri KR, et al. Time course of symptomatic orthostatic hypotension and urinary incontinence in patients with postmortem confirmed parkinsonian syndromes: a clinicopathological study. J Neurol Neurosurg Psychiatry. BMJ Publishing Group; 1999;67:620–623.
Boeve BF, Silber MH, Ferman TJ, Lin SC, Benarroch EE, Schmeichel AM, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013;14:754–62.
Tomše P, Jensterle L, Rep S, Grmek M, Zaletel K, Eidelberg D, et al. The effect of 18F-FDG-PET image reconstruction algorithms on the expression of characteristic metabolic brain network in Parkinson’s disease. Phys Med. 2017;41:129–35.
Tomše P, Peng S, Pirtošek Z, Zaletel K, Dhawan V, Eidelberg D, et al. The effects of image reconstruction algorithms on topographic characteristics, diagnostic performance and clinical correlation of metabolic brain networks in Parkinson’s disease. Phys Medica. 2018;52:104–12.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
DE serves as a member of scientific advisory board of Ovid Therapeutics (receives personal fees) and has served on the scientific advisory board of The Michael J. Fox Foundation (has received honoraria); is a consultant to MeiraGTx (has received fees); is listed as coinventor of patents for markers for use in screening patients for nervous system dysfunction as well as a method and apparatus for using same (without financial gain); has received research support from the National Institutes of Health (NINDS, NIAID) and The Michael J. Fox Foundation for Parkinson’s Research. The other authors have no relevant conflicts of interest to declare.
Informed consent
Written informed consent was obtained from the participating subjects.
Ethical approval
All procedures performed in the study involving human participants were in accordance with the ethical standards of the national research committee (The National Medical Ethics Committee of the Republic of Slovenia, 0120-351/2015-2, KME 70/07/15) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neurology
Electronic supplementary material
ESM 1
(PDF 333 kb)
Rights and permissions
About this article

Cite this article
Rus, T., Tomše, P., Jensterle, L. et al. Differential diagnosis of parkinsonian syndromes: a comparison of clinical and automated - metabolic brain patterns’ based approach. Eur J Nucl Med Mol Imaging 47, 2901–2910 (2020). https://doi.org/10.1007/s00259-020-04785-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-020-04785-z