
Abstract
Purpose
Niemann-Pick type C (NPC) is a cholesterol storage disease characterized by disruption in the endosomal–lysosomal transport system that leads to the accumulation of cholesterol and glycolipids in lysosomes. Developmental cognitive delay and progressive motor and cognitive impairment are characteristic of the disease. Tau accumulation has been reported in some NPC patients. We investigated the presence of tau and Aβ-amyloid deposits in a group of NPC patients and for comparison in age-matched healthy controls (HC).
Methods
Eight NPC patients and seven HC were included in the study. Participants underwent tau imaging with 18F-AV1451 and amyloid imaging with 11C-PiB. Both 18F-AV1451 and 11C-PiB standardized uptake value ratios were generated using the cerebellar cortex as the reference region. Associations between imaging results, and clinical and neurocognitive parameters were assessed through nonparametric analyses.
Results
All participants were Aβ-negative. Four NPC patients presented with high tau burden in the brain. A 21-year-old female patient and a 40-year-old male patient showed high neocortical tau burden in a pattern different from that observed in patients with Alzheimer’s disease, while the same 40-year-old male patient, a 40-year-old female patient and a 50-year-old female patient showed high regional tau burden in the mesial temporal cortex. Spearman’s correlation analysis showed an association between tau burden in the mesial temporal lobe and age (p = 0.022), and age at symptom onset (p = 0.009), and between frontotemporal tau and duration of symptoms (p = 0.027). There were no correlations between global and regional tau and cognitive parameters.
Conclusion
Four of eight NPC patients showed tau deposition in the brain. The results of our exploratory study suggest that while tau deposits do not affect cognitive performance, tau deposits are associated with measures of disease onset and progression. Further studies in a larger cohort of NPC patients are needed to confirm these initial findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Niemann-Pick type C (NPC) disease is an autosomal recessive neurovisceral and neurodegenerative disorder that involves alterations in intracellular sterol trafficking. NPC disease is caused by mutations in the NPC1 gene (in 95% of patients) and the NPC2 gene (in 5%) [1, 2], and affects an estimated 1 in 90,000 live births [3]. Impairment of the function of NPC1/2 alters sterol cycling between the late endosome and lysosome, with accumulation of glycosphingolipids, especially GM2 and GM3 gangliosides, in neurons and unesterified cholesterol and sphingomyelin in the liver. NPC presents with significant clinical heterogeneity across the lifespan, and may present with visceral symptoms (neonatal jaundice and hepatosplenomegaly), neurological impairment (ataxia, dystonia, vertical saccadic palsy, and cognitive impairment) across the lifespan [4], and neuropsychiatric symptoms (particularly psychosis) in adolescents and young adults.
Loss of NPC1 function alters neuronal morphology, with abnormal neuronal storage of lipids, neuroaxonal dystrophy with ectopic dendritogenesis and formation of meganeurites, which precede axonal and neuronal degeneration, typical findings in lysosomal storage diseases [5]. Purkinje cells in the cerebellum, basal ganglia and thalamus are the most vulnerable neurons to NPC-induced neuronal dysfunction, and these grey matter regions are the first affected with hippocampal and cortical regions affected later [6, 7], Affected neurons often show ectopic dendritogenesis with stunted dendrites and greatly reduced dendritic arborization [8] as a result of altered phosphorylation of the microtubule-associated protein MAP2 which results in dendritic microtubule depolymerization [9] and a reduced availability of arborization-promoting neurosteroids secondary to cholesterol unavailability [10]. In adult patients regional neuronal loss in subcortical regions and widespread white matter changes can be detected by modern in vivo neuroimaging techniques [11,12,13].
Paired helical filament-containing neurofibrillary tangles (NFTs), one of the key pathological hallmarks of Alzheimer’s disease (AD), also appear to be a significant additional feature of NPC. They were first described in 1978 in a neuropathological study of a 29-year-old woman with “neurovisceral lipidosis”, now thought to have been NPC [14]. The pathological findings included distended neurons and axons in the frontotemporal cortex, hippocampal complex and basal ganglia. A number of subsequent case series demonstrated that NFTs occur in both paediatric and adult NPC patients, and are immunohistochemically and ultrastructurally identical to those seen in AD [15,16,17]. NFTs have been seen in patients with NPC as young as 4 years old [18]. In contrast to AD, NFTs in NPC are found in subcortical structures including the thalamus, basal ganglia and hippocampus, more than in neocortical regions such as the frontal and temporal cortices. Unlike in AD, NFTs are rarely seen in the parietal and occipital regions in patients with NPC [15,16,17, 19]. NFTs also tend to be colocated in neurons most affected by abnormal cholesterol and sphingolipid storage [20]. Further contrasting with AD, dense core Aβ-amyloid plaques are not seen in NPC patients, in spite of altered Aβ42 processing [16]. It has been suggested that the lack of cortical plaques may result in less neocortical spread of NFTs in NPC than in AD [20]. However, in spite of the fact that the most fulminant neuronal loss in NPC occurs in the cerebellum, NFTs are not present in the cerebellum [16, 17, 21], which may reflect the fact that tau levels are up to fivefold lower in Purkinje neurons in this brain region [21].
The advent of noninvasive in vivo Aβ-amyloid and tau imaging with positron emission tomography (PET) has led to a deeper insight into the pathophysiology of the deposition of these proteinopathies in the brain [22]. These biomarkers allow better disease staging, as well as assessment of the relationship with other cognitive, imaging and fluid biomarkers, that are crucial for patient selection as well as an outcome measure in disease-specific trials [22]. To date, no studies have investigated in vivo Aβ-amyloid and tau deposition in NPC. In the current study, we assessed a group of young adult NPC patients to determine the presence or absence of tau and/or Aβ-amyloid deposition in the brain, as well as their relationship to clinical and cognitive variables.
Materials and methods
Eight NPC patients (four women, four men; median age 33.5 years, IQ range 22–40, age range 18–50 years) and seven healthy controls (HC; five women, two men; median age 29.4 years IQ range 24–47, age range 21–49 years) were included in the study, including two sibling pairs with identical mutation status (Table 1). AV1451 scans of the three youngest HC were provided by Avid Radiopharmaceuticals. Written informed consent was obtained from all participants and/or their carers. Approval for the study was obtained from the Human Research Ethics Committee at Austin Health. The clinical features of each NPC patient are shown in Table 1. All NPC patients underwent neurocognitive assessment using the Neuropsychiatry Unit Cognitive Assessment Tool (NUCOG) that evaluates five cognitive domains (attention, memory, language, executive and visuospatial function). A total score of <80/100 is considered abnormal [23]. All patients were rated at the time of scanning on the NPC illness rating scale developed by Iturriaga et al. [24].
Seven NPC patients underwent a 3D T1 MPRAGE MRI scan. One of these MRI scans was not suitable for volumetric analysis due to movement artefacts. The other NPC patient underwent a clinical T1 MRI scan. No partial volume correction of the PET data was performed. Tau imaging emission scans were obtained between 80 and 100 min after injection of 200 MBq of 18F-AV1451, while Aβ imaging scans were obtained with either 11C-PiB (40–70 min after injection) or 18F-florbetapir (50–70 min after injection). Global and regional tau (18F-AV1451) burdens are expressed as standardized uptake value ratios (SUVR) using the cerebellar cortex as the reference region. Z-scores were generated using the control group, and were examined using the neocortical and MeTeR regional scale, where Me represents the mesial temporal region (comprising the hippocampus, parahippocampus, amygdala and entorhinal cortex), T represents the temporoparietal region (comprising the inferior and middle temporal lobes, supramarginal and angular gyrus, inferior and superior parietal, lateral occipital and posterior cingulate/precuneus), and R represents the rest of the neocortex (comprising the dorsolateral and ventrolateral prefrontal, orbitofrontal, superior temporal gyrus and anterior cingulate) [25] similar to Braak stages I/II, III/IV, and V–VI, respectively [26].
The SUVRCbCtx cut-off value for AV1451 was established as a z-score of 2. Nonparametric (Wilcoxon) tests were used for comparison of groups. Spearman correlations were applied to assess the relationship between tau burden, and cognitive and clinical parameters. No adjustment for multiple comparisons was performed.
Results
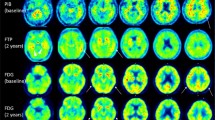
All participants were Aβ-negative according to the Aβ PET scan. Two NPC patients (patients 5 and 8) showed high overall tau burden in the brain. Two NPC patients (patients 3 and 5) showed high neocortical and Te and R tau burdens (z-score greater than 2; Table 2). Half of the NPC patients showed high tau burden in the Me (Table 2, Fig. 1). The high neocortical tau burden in patients 3 and 5 also showed a pattern different from that usually observed in the amnestic presentation of AD (Fig. 2). However, not all NPC patients showed a detectable tau burden (Fig. 2). Another 40-year-old female patient (patient 8) showed a very high regional tau burden in the mesial temporal region, with incipient tracer retention in the temporoparietal cortices (Fig. 2). Probably due to the young age of the cohort, no age-related off-target AV1451 retention was observed in the basal ganglia or choroid plexus.

Neocortical and regional tau burden in NPC patients (NPC) and healthy controls (HC). The z-score scatterplots for neocortical and regional (Me mesial temporal, Te temporoparietal, R rest of neocortex) tau burden show high tau burden in some NPC patients. High neocortical, Te and R tau burdens were observed in patients 3 and 5, and high Me tau burden was observed in patients 5, 6 and 8. The horizontal dashed lines indicate a z-score of 2, used as a cut-off value between high and low tau burdens

Average surface projections of 18F-AV1451 SUVR in healthy controls (HC), and individual 18F-AV1451 SUVR surface projections in NPC patients (NPC1-8). When present (NPC3, NPC5, NPC6 and NPC8), the pattern of 18F-AV1451 retention with high retention in the frontal, mesial temporal cortex, inferior and middle temporal gyri, supra-marginal gyrus and orbitofrontal regions is somewhat different from the typical mesial temporal, temporoparietal and posterior cingulate tracer retention pattern observed in amnestic Alzheimer’s disease
Nonparametric analysis in NPC patients showed a significant correlation between tau burden in Me and age (Spearman’s ρ = 0.78, p = 0.022), and age at symptom onset (Spearman’s ρ = 0.84, p = 0.009), and between tau burden in R and duration of symptoms (Spearman’s ρ = −0.77, p = 0.027). However, there were no significant correlations between global and regional tau burden and cognitive parameters.
Discussion
This study identified significant tau pathology in four of eight adult patients with NPC, in a distribution that varied between patients and which differed from the pattern typically observed in patients with amnestic sporadic AD [27]. The other two main findings of this study were that tau deposition was significantly associated with illness duration and that no patient exhibited significant Aβ-amyloid pathology. These findings suggest that tau aggregates may play a role in the pathology of NPC.
Detectable tau pathology was not observed in all NPC patients, even those with significant cognitive impairment and duration of neurological symptoms. One possible explanation for this is that the regional load of “detectable” NFTs in NPC is relatively low and variable in comparison to that found in AD, even in NPC patients of similar ages [17]. In vivo binding of 18F-AV1451 resembles the known post-mortem NFT load and Braak staging in NPC patients, and is most strongly detectable in patients with Braak stage III and beyond [28]. It should be noted that the pattern of 18F-AV1451 retention in NPC patients differed from that observed in patients with the typical amnestic presentation of AD, who tend to show higher 18F-AV1451 retention predominantly in the medial temporal and inferior temporal lobe regions, whereas patients with early-onset disease tend to show predominantly more neocortical impairment affecting the temporal and parietal regions, corresponding to Braak stages V and VI [29]. There are no comparative studies that have compared tangle density between NPC and AD patients, although reports do suggest that some NPC patients have sparse populations of NFTs [15,16,17]. Thus, while a number of adult NPC patients may show an NFT distribution pattern akin to Braak stage III in AD [30], it may be that NFT density in some patients is insufficient to show a significant PET signal. There have also been suggestions that NFTs may accumulate in an age-related fashion [18], and our NPC cohort was significantly younger than a typical AD cohort.
Additionally, it may be that in some NPC patients, tangle morphology differs from the classical flame-like tangles seen in AD, and thus affects the binding of 18F-AV1451 that is known to bind strongly to some conformations, but weakly to others [31]. Comparative autoradiographic and brain homogenate studies with 18F-AV1451 between AD and NPC patients are needed to account for this discrepancy, to ascertain the relative binding of the tracer to tau in each condition, and if it is related to binding affinity, variability in tangle load or tau morphology.
Unlike in AD, none of our patients showed significant Aβ-amyloid pathology, suggesting a different pathophysiology in NPC from that in AD. Many hypotheses have been proposed to explain different underlying pathophysiological mechanisms in NPC, such as disturbed cholesterol metabolism. It has been demonstrated that induced cholesterol deficiency in cultured neurons results in microtubule depolymerization and hyperphosphorylation of tau [32], and in NPC abnormal cholesterol trafficking and metabolism may be responsible for the activation of the mitogen-activated protein kinase (MAPK) signalling pathway resulting in tau phosphorylation [33], although cdc2/cyclin B kinase-mediated phosphorylation may also play a role [18]. Furthermore, tangle-bearing neurons in NPC tend to be those neuronal populations enriched with free cholesterol and other storage material [17, 34].
The relationship between tau burden and symptom duration was notable. This finding suggests that a detectable tau burden in the adult NPC group may be associated with a more rapidly progressing form of the disease in this subgroup of patients. This appeared to be a function of tau, rather than age, as this was observed in patients both younger and older than 35 years.
Although no significant associations were observed between tau burden and cognition as assessed using the NUCOG assessment tool, a strong trend was observed between executive function and temporoparietal tau. This might suggest that pathology in the temporoparietal region disrupts one “node” of corticocortical connectivity that subserves a range of executive functions as indexed by the NUCOG assessment tool. Cortical tau has been shown to be associated with objective and subjective cognitive impairment in AD [35, 36]. Our trend was most strongly seen in executive functioning rather than memory, indicating that the frontal tau burden is probably higher in NPC patients than in AD patients [20]. Notably, in patients with early-onset AD, 18F-AV1451 binding in temporoparietal regions is associated with impairment in the performance of executive, attention and spatial tasks more notably than memory function [37]. A larger cohort of NPC patients is needed to clarify the potential relationship between tau burden and cognition.
The role of tau pathology is often overlooked in NPC research. Tau plays a central role in microtubule stabilization and polymerization in axons, and pathological hyperphosphorylation of tau decreases its affinity for microtubules with increased propensity to aggregate to form NFTs. Axonal changes are a hallmark neuropathological feature of NPC [5], and we have previously shown that widespread axonal changes appear to occur in the brain of adult NPC patients, and can be demonstrated in vivo using diffusion imaging [12], Total tau levels are also elevated in the cerebrospinal fluid of NPC patients, and respond to treatment with the illness-modifying drugs miglustat and cyclodextrin, indicating that axonal disruption is a core pathological feature of the illness [38,39,40]. Current experimental data suggest that tau pathology in NPC is due to a combination of increased activation of tau kinases, such as MAPK [33] and cyclin-dependent kinases 4 and 5 (CDK4 and CDK5) [18, 41], and decreased activity of tau phosphatases (e.g. protein phosphatase 2A) [33]. This raises the intriguing possibility that compounds that modulate the activity of tau kinases and phosphatases may be of therapeutic benefit by restoring the equilibrium of tau phosphorylation and dephosphorylation.
There were limitations to this study. The exploratory nature of the study is reflected in the small number of NPC patients and age-matched HCs that precludes the extension of these findings to all individuals affected by this disease. More studies in younger normal populations are required to refine global and regional thresholds to confidently allow identification of high levels of tau deposition.
Conclusion
We have demonstrated that tau burden is detectable in vivo in adult NPC patients using 18F-AV1451 that has been shown to bind to paired helical filaments in patients with other tauopathies. In contrast to AD, tau deposition in NPC does not seem to be related to Aβ deposition. The pattern of tauopathy in NPC patients seems to be different from that observed in patients with typical late-onset AD and may reflect the younger age of adult NPC patients. That not all patients show tau deposition as assessed by 18F-AV1451 is concordant with post-mortem findings, but it may also suggest a subthreshold amount of tau in the brain, or differences in the conformation of the tau deposits. Our findings suggest that detectable tau burden in these patients may reflect a particular subtype of the illness associated with a more rapid disease progression.
References
Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–31.
Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290(5500):2298–301. https://doi.org/10.1126/science.290.5500.2298.
Wassif CA, Cross JL, Iben J, Sanchez-Pulido L, Cougnoux A, Platt FM, et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med. 2016;18(1):41–8. https://doi.org/10.1038/gim.2015.25.
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106(3):330–44. https://doi.org/10.1016/j.ymgme.2012.03.012.
Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685(1-3):48–62. https://doi.org/10.1016/j.bbalip.2004.08.011.
March P, Thrall M, Brown D, Mitchell T, Lowenthal A, Walkley S. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol. 1997;94:164–72.
Ong W, Kumar U, Switzer R, Sidhu A, Suresh G, Hu C, et al. Neurodegeneration in Niemann-Pick type C disease mice. Exp Brain Res. 2001;141:218–31.
Paul C, Boegle A, Maue R. Before the loss: neuronal dysfunction in Niemann-Pick type C disease. Biochim Biophys Acta. 2004;1685:63–76.
Fan Q-W, Yu W, Gong J-S, Zou K, Sawamura N, Senda T, et al. Cholesterol-dependent modulation of dendrite outgrowth and microtubule stability in cultured neurons. J Neurochem. 2002;80:178–90.
Sakamoto H, Ukena K, Tsutsu K. Effects of progesterone synthesized de novo in the developing Purkinje cell on its dendritic growth and synaptogenesis. J Neurosci. 2001;16:6221–32.
Walterfang M, Abel LA, Desmond P, Fahey MC, Bowman EA, Velakoulis D. Cerebellar volume correlates with saccadic gain and ataxia in adult Niemann-Pick type C. Mol Genet Metab. 2013;108(1):85–9. https://doi.org/10.1016/j.ymgme.2012.11.009.
Walterfang M, Fahey M, Desmond P, Wood A, Seal ML, Steward C, et al. White and gray matter alterations in adults with Niemann-Pick disease type C: a cross-sectional study. Neurology. 2010;75(1):49–56. https://doi.org/10.1212/WNL.0b013e3181e6210e.
Walterfang M, Patenaude B, Abel LA, Kluenemann H, Bowman EA, Fahey MC, et al. Subcortical volumetric reductions in adult Niemann-Pick disease type C: a cross-sectional study. AJNR Am J Neuroradiol. 2013;34(7):1334–40. https://doi.org/10.3174/ajnr.A3356.
Horoupian DS, Yang SS. Paired helical filaments in neurovisceral lipidosis (juvenile dystonic lipidosis). Ann Neurol. 1978;4(5):404–11. https://doi.org/10.1002/ana.410040504.
Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, et al. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol. 1995;90(6):547–51.
Love S, Bridges LR, Case CP. Neurofibrillary tangles in Niemann-Pick disease type C. Brain. 1995;118(Pt 1):119–29.
Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D'Agostino AN, et al. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995;89(3):227–38.
Zhang M, Li J, Chakrabarty P, Bu B, Vincent I. Cyclin-dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann-Pick type C mice. Am J Pathol. 2004;165(3):843–53.
Chiba Y, Komori H, Takei S, Hasegawa-Ishii S, Kawamura N, Adachi K, et al. Niemann-Pick disease type C1 predominantly involving the frontotemporal region, with cortical and brainstem Lewy bodies: an autopsy case. Neuropathology. 2014;34(1):49–57. https://doi.org/10.1111/neup.12047.
Bergeron D, Poulin S, Laforce R Jr. Cognition and anatomy of adult Niemann-Pick disease type C: insights for the Alzheimer field. Cogn Neuropsychol. 2018;35(3-4):209–22.
Bu B, Klunemann H, Suzuki K, Li J, Bird T, Jin LW, et al. Niemann-Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol Dis. 2002;11(2):285–97.
Villemagne VL, Doré V, Burnham SC, Masters CL, Rowe CC. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol. 2018;14(4):225–36.
Walterfang M, Siu R, Velakoulis D. The NUCOG: validity and reliability of a brief cognitive screening tool in neuropsychiatric patients. Aust N Z J Psychiatry. 2006;40(11–12):995–1002. https://doi.org/10.1111/j.1440-1614.2006.01923.x.
Iturriaga C, Pineda M, Fernandez-Valero EM, Vanier MT, Coll MJ. Niemann-Pick C disease in Spain: clinical spectrum and development of a disability scale. J Neurol Sci. 2006;249(1):1–6. https://doi.org/10.1016/j.jns.2006.05.054.
Villemagne V, Dore V, Bourgeat P, Burnham S, Mulligan R, Laws S, et al. The Tau MeTeR composites for the generation of continuous and categorical measures of tau deposits in the brain. J Mol Med Ther. 2017;1(1):25–9.
Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18(4):351–7.
Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016;139(Pt 5):1551–67. https://doi.org/10.1093/brain/aww027.
Marquie M, Siao Tick Chong M, Anton-Fernandez A, Verwer EE, Saez-Calveras N, Meltzer AC, et al. [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging. Acta Neuropathol. 2017;134(4):619–28. https://doi.org/10.1007/s00401-017-1740-8.
Scholl M, Ossenkoppele R, Strandberg O, Palmqvist S, Swedish Bio F, Jogi J, et al. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer's disease. Brain. 2017;140(9):2286–94. https://doi.org/10.1093/brain/awx171.
Zhang M, Wang X, Jiang F, Wang W, Vincent I, Bu B. Mitotic epitopes are incorporated into age-dependent neurofibrillary tangles in Niemann-Pick disease type C. Brain Pathol. 2010;20(2):367–77.
Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain. 2017;140(3):764–80. https://doi.org/10.1093/brain/aww339.
Fan QW, Yu W, Senda T, Yanagisawa K, Michikawa M. Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J Neurochem. 2001;76(2):391–400.
Sawamura N, Gong JS, Chang TY, Yanagisawa K, Michikawa M. Promotion of tau phosphorylation by MAP kinase Erk1/2 is accompanied by reduced cholesterol level in detergent-insoluble membrane fraction in Niemann-Pick C1-deficient cells. J Neurochem. 2003;84(5):1086–96.
Distl R, Meske V, Ohm TG. Tangle-bearing neurons contain more free cholesterol than adjacent tangle-free neurons. Acta Neuropathol. 2001;101(6):547–54.
Pontecorvo MJ, Devous MD Sr, Navitsky M, Lu M, Salloway S, Schaerf FW, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140(3):748–63. https://doi.org/10.1093/brain/aww334.
Buckley RF, Hanseeuw B, Schultz AP, Vannini P, Aghjayan SL, Properzi MJ, et al. Region-specific association of subjective cognitive decline with tauopathy independent of global beta-amyloid burden. JAMA Neurol. 2017;74(12):1455–63.
Cho H, Chooi J, Lee S, Hwang M, Ryu Y, Lee M, et al. Tau burden and cognition in early-onset versus late-onset Alzheimer’s disease spectrum. Alzheimers Dement. 2016;12(7):S711–2.
Mattsson N, Zetterberg H, Bianconi S, Yanjanin NM, Fu R, Mansson JE, et al. Gamma-secretase-dependent amyloid-beta is increased in Niemann-Pick type C: a cross-sectional study. Neurology. 2011;76(4):366–72. https://doi.org/10.1212/WNL.0b013e318208f4ab.
Mattsson N, Zetterberg H, Bianconi S, Yanjanin NM, Fu R, Mansson JE, et al. Miglustat treatment may reduce cerebrospinal fluid levels of the axonal degeneration marker tau in Niemann-Pick type C. JIMD Rep. 2012;3:45–52. https://doi.org/10.1007/8904_2011_47.
Matsuo M, Shraishi K, Wada K, Ishitsuka Y, Doi H, Maeda M, et al. Effects of intracerebroventricular administration of 2-hydroxypropyl-beta-cyclodextrin in a patient with Niemann-Pick type C disease. Mol Genet Metab Rep. 2014;1:391–400. https://doi.org/10.1016/j.ymgmr.2014.08.004.
Bu B, Li J, Davies P, Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci. 2002;22(15):6515–25.
Acknowledgments
We thank Avid Radiopharmaceuticals for providing AV1451 precursor and standard, especially Drs. Michael Pontecorvo and Michael Devous, for kindly providing 18F-AV1451 images of three young adults used for age-matched comparison with young NPC patients. We also thank Dr. Graeme O’Keefe, Dr. Gordon Chan, Dr. Kenneth Young, Dr. Sylvia Gong, Mrs. Denise El-Sheikh; and the Brain Research Institute for their assistance with this study.
Funding
This work was supported in part by the Austin Hospital Medical Research Foundation. The funding source was not involved in the study design, in the collection, analysis and interpretation of the data, in the writing of the report, or in the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
V.L.V. and M.W. conceived and designed the research. V.L.V., M.W., D.V., V.D., S.B., C.L.M. and C.C.R. performed the research and participated in the drafting of the work and revising it critically for important intellectual content. V.L.V. and M.W. wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Conflicts of interest
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Villemagne, V.L., Velakoulis, D., Doré, V. et al. Imaging of tau deposits in adults with Niemann-Pick type C disease: a case-control study. Eur J Nucl Med Mol Imaging 46, 1132–1138 (2019). https://doi.org/10.1007/s00259-019-4273-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-019-4273-7