
Abstract
Purpose
The endoplasmic reticulum (ER) contains hexose-6P-dehydrogenase (H6PD). This enzyme competes with glucose-6P-phosphatase for processing a variety of phosphorylated hexoses including 2DG-6P. The present study aimed to verify whether this ER glucose-processing machinery contributes to brain FDG uptake.
Methods
Effect of the H6PD inhibitor metformin on brain 18F-FDG accumulation was studied, in vivo, by microPET imaging. These data were complemented with the in vitro estimation of the lumped constant (LC). Finally, reticular accumulation of the fluorescent 2DG analogue 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2NBDG) and its response to metformin was studied by confocal microscopy in cultured neurons and astrocytes.
Results
Metformin halved brain 18F-FDG accumulation without altering whole body tracer clearance. Ex vivo, this same response faced the doubling of both glucose consumption and lactate release. The consequent fall in LC was not explained by any change in expression or activity of its theoretical determinants (GLUTs, hexokinases, glucose-6P-phosphatase), while it agreed with the drug-induced inhibition of H6PD function. In vitro, 2NBDG accumulation selectively involved the ER lumen and correlated with H6PD activity being higher in neurons than in astrocytes, despite a lower glucose consumption.
Conclusions
The activity of the reticular enzyme H6PD profoundly contributes to brain 18F-FDG uptake. These data challenge the current dogma linking 2DG/FDG uptake to the glycolytic rate and introduce a new model to explain the link between 18-FDG uptake and neuronal activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the seminal work by Sokoloff et al. [1], the tight and local connection between glucose consumption and neuronal activation has been exploited to map brain response to different stimulations using 14C-2-deoxyglucose (2DG) autoradiography. The robustness of this method allowed its translation to humans, with PET/CT imaging of 18F-fluorodeoxyglucose (FDG) that currently represents a clinical standard in many neurodegenerative disorders [2]. The underlying rationale relies on the assumption that both 2DG and FDG compete with glucose for transport across both blood-brain barrier and cell membrane, as well as for hexokinase (HK) catalyzed phosphorylation [3, 4]. However, differently from glucose-6-phosphate (G6P), both 2DG-6-phosphate (2DG6P) and FDG-6-phosphate (FDG6P) cannot be further degraded through the two known pathways, glycolysis and pentose phosphate pathway (PPP) [4]. Thus, neglecting tracer loss caused by the slow hexose dephosphorylation [1], normalizing tissue uptake for blood tracer availability permits researchers to noninvasively estimate regional cerebral metabolic rate of glucose (CMRGlu*).
Although this method represents the basis for most of our current knowledge about the link between local neuronal activation and glucose metabolism in different disorders, its validity has not been conclusively defined. Indeed, a significant fraction of intracellular 2DG has been found not to be processed by HK [5], while magnetic resonance spectroscopy has documented a significant degradation of FDG6P [6,7,8], suggesting that the link between glucose consumption and 2DG-FDG uptake might be relatively looser than conventionally assumed. In agreement with this concept, our group recently documented in a broad panel of cancer cell lines that FDG uptake is strictly dependent upon the activity of hexose-6-phosphate dehydrogenase (H6PD) [9], the autosomic counterpart of G6P-dehydrogenase (G6PD), able to dehydrogenate a large number of hexoses, including 2DG6P, within the endoplasmic reticulum (ER) [10, 11].
To define the role of H6PD in brain FDG uptake, this study evaluated the response of whole brain, primary neurons and primary astrocytes to metformin (MTF). This anti-diabetic drug crosses the blood-brain barrier [12], is a powerful inhibitor of H6PD [9] and mitochondrial complex I function [13], eventually accelerating glycolytic flux in both neurons and astrocytes [14, 15].
Material and methods
Animal models
Six-week-old BALB/c mice were purchased from Charles River Laboratories (Italy) and housed under specific pathogen-free conditions. Fifteen ‘control’ mice were kept under standard conditions, and 15 ‘treated’ mice received MTF (Sigma Aldrich) diluted in their drinking water at a concentration of 3 mg/mL, accounting for a dose of approximately 750 mg/Kg/d [9].
Experimental micro-PET imaging
In vivo imaging was performed according to a protocol validated in our lab [9] in 12 mice per group. Mice were weighed and anaesthesia was induced by intra-peritoneal administration of ketamine/xylazine (100 and 10 mg/kg, respectively). Serum glucose level was tested and animals were positioned on the bed of a dedicated micro-PET system (Albira, Bruker, US). A dose of 3-4 MBq of FDG was then injected through a tail vein, soon after start of a 50 min list-mode acquisition.
PET data were reconstructed using a maximal likelihood expectation maximization method (MLEM) using the following framing rate: 10 × 15, 5 × 30, 2 × 150, 6 × 300, 1 × 600 s). An experienced observer, unaware of the experimental type of analysed mouse, identified a volume of interest (VOI) in the left ventricular chamber to plot the time-concentration curve. Whole body FDG clearance (in ml × min−1) was calculated using the conventional stochastic approach as the ratio between injected dose and integral of input function from 0 to infinity, fitting the last 20 min with a mono-exponential function [16]. CMRGlu* (in nMol × min−1 × g−1) was estimated in the whole brain according to Gjedde-Patlak [17] graphical analysis using the routine of dedicated software (PMOD, Zurich, Switzerland) without correction for the lumped constant (LC). Any interference of MTF on FDG exchange between erythrocytes and plasma was excluded according to the procedure described in the supplementary material and supplementary Fig. 1.
Ex vivo measurement of brain metabolism
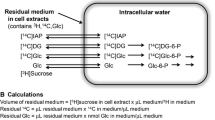
Ex vivo evaluation was completed using a dedicated instrument (LigandTracer White, Ridgeview, SE) that consists of a beta-emission detector and a rotating platform harbouring a standard Petri dish. The rotation axis is inclined at 30° from the vertical, so that the organ alternates its position from the nadir (for incubation) to the zenith (for counting) every minute (Suppl Fig. 1). Time-activity curves are thus obtained by subtracting decay-corrected background counting rate from the corresponding target value [16].
For the ex vivo study, brains of three control and ‘treated’ mice, respectively, were harvested soon after sacrifice. Slices 300 μM thick were stuck in the outer ring of a Petri dish with octyl-cyanoacrylate (Dermabond, Ethicon, US) and covered with 2 mL solution collected from an input vial containing 3 mL of Dulbecco’s Modified Eagle’s Medium (DMEM) containing glucose and FDG at the concentration of 11.1 mM and 1 MBq/mL, respectively. For MTF-brains, incubation medium was enriched with MTF 10 mM. At the end of the experiment, an aliquot of 0.5 mL was sampled both from the input vial and from the Petri dish (output). In both samples, radioactivity concentration was measured using a dose calibrator with an activity resolution <10 KBq (Capintec CRC55). FDG uptake was thus expressed
where Ainput and Aoutput represent activity in MBq in the input and in the output, respectively. Similarly, glucose consumption (CMRGlu) and lactate production were measured as:
where “metabolite change” indicates either lactate production (with inverted sign) or CMRGlu, while Cinput and Coutput represent the corresponding mM concentrations, and 2 × 10−3 represents the volume of used medium. The possible interference of MTF on the measurement of glucose concentration was excluded following to the procedure described in the supplementary material.
The availability of CMRGlu and fractional FDG uptake thus permitted us to estimate LC [1, 5, 18,19,20,21,22] defined as:
where λ indicates the ratio between distribution volumes of glucose and FDG and thus GLUT contribution, \( \frac{1}{\phi } \) represents the different G6Pase affinities for the two hexoses, while the last two ratios refer to the kinetics of HK-catalysed phosphorylation for glucose (plain text) and 2DG (flagged with *, as in the original Sokoloff formulation). Both Vmax (the maximum rate achieved by the system) and Km (the Michaelis-Menten constant indicating the substrate concentration at which the reaction rate is Vmax/2) were determined, at pH 7.4 and 25 °C, by coupling hexose phosphorylation to the reduction of NADP, recording the change in absorbance at 340 nm. Km values were determined by Lineweaver-Burk double reciprocal plots. Substrate concentrations were 0.05, 0.1, 1, 5 and 200 mM for glucose and 0.3, 5, 50, 100 and 200 mM for 2DG. To avoid the substrate selectivity, G6PD was substituted with H6PD, as an enzyme able to process both hexoses.
Autoradiography
Autoradiography was performed according to our standard procedure [16] as described in the supplementary material.
Co-localization experiments
For these experiments, cultured astrocytes and neurons were incubated for 20 min in a medium containing glucose (25 mM), the 2D analogue 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2NBDG) (50 microM) and the ER-Tracker™ Red (1 microM), (Molecular Probes, InVitrogen, Eugene, OR). After extensive washing, cellular fluorescence was measured on the SP2-AOBS confocal microscope (Leica Microsystems, Mannheim, Germany) using a 488 nm laser for 2NBDG excitation (emission detection range 500-560 nm) and a 546 nm laser for ER-Tracker excitation (emission detection range 600-700 nm). Six to eight randomly selected fields containing at least 8–10 cells were analysed in three independent samples for each treatment. Co-localization of the two signals was tested according to Costes method (ImageJ software, NIH) [23] that also provided average signal intensity in all pixels above the automatically identified threshold.
Preparation of primary cell cultures and western blot analysis
Procedures for cultures of primary neurons and astrocytes, western blot and real-time PCR analyses are described in the supplementary material.
Spectrophotometric assay
Enzymatic assays were performed in a double beam spectrophotometer (UNICAM UV2, Analytical S.n.c., Italy) [9].
HK, H6PD and G6PD activities were assayed following reduction of NADP at 340 nm. Phosphofructokinase (PFK) activity was assayed following oxidation of NADH at 340 nm. Complex I activity was assayed following the reduction of ferrocyanide (FeCN), at 420 nm.
The following assays solutions were used: HK: Tris-HCl-pH 7.4-100 mM (TRIS7.4), MgCl2 2 mM, glucose 200 mM, ATP 1 mM, NADP 0.5 mM, H6PD (Sigma-Aldrich); H6PD: TRIS7.4, 2DG6P 10 mM, NADP 0.5 mM; G6PD: TRIS7.4, G6P 10 mM, NADP 0.5 mM; PFK: Tris-HCl pH 8100 mM, MgCl2 2 mM, KCl 5 mM, fruttose-6-phpsphate 2 mM, ATP 1 mM, phosphoenolpyruvate (PEP) 0.5 mM, NADH 0.2 mM, pyruvate kinase (PK)/lactate dehydrogenase (LDH) (Sigma-Aldrich); Complex I: TRIS7.4, NADH 0.6 mM, antimycin 50 μM, FeCN 0.8 mM.
Glucose and lactate in medium were assayed following the reduction of NADP and NAD+ at 340 nm, respectively. The following solutions were used: glucose: TRIS7.4, MgCl2 0.2 mM, ATP 2 mM, HK/G6PD (Sigma-Aldrich); lactate: Tris-HCl-pH 9100 mM, NAD+ 5 mM, LDH (Sigma-Aldrich).
G6Pase activity on G6P or 2DG6P was measured at 660 nm, following the inorganic phosphate production [24], using 150 μg brain proteins. The assay solution contained: G6P or 2DG6P 40 mM, tris-maleic acid pH 6.5 60 mM. Following an incubation at 37 °C for 15 min, the reaction was blocked with tri-chloro-acetic acid 2%. Samples were centrifuged at 14000 rpm for 2 min and the supernatant was added with: H2SO4 350 mM, ammonium molybdate 350 mM, Fiske’s reactive.
Oxygen consumption
Oxygen consumption was measured using an amperometric oxygen electrode (Microrespiration, Unisense A/S, Århus, Denmark) in a closed chamber (volume 1.7 ml); magnetically stirred at room temperature. 100,000 cells were used during the experiments. The intact cells were centrifuged to remove the growth medium at 1000 g for 2 min. The pellet was suspended in the respiration buffer composed of KCl (5 mM) NaCl (0.137 M), Na2HPO4 (0.7 mM) and TRIS7.4 (25 mM). Glucose (20 mM) or 2DG (20 mM) were used as substrates. Absent response to ADP (0.5 mM) confirmed cell integrity in all experiments.
Statistical analysis
The data are presented as mean ± standard deviation (SD). For comparison between different groups, the Null hypothesis was tested by Student’s t test for unpaired data. Statistical significance was considered for p values p < 0.05. All analyses were performed using the SPSS software package, 20.0.0 release (IBM, Armonk, NY).
Results
Metabolic response to metformin in the whole body and brain
Treatment was completed in all 12 animals without side effects. As shown in Table 1 and in Fig. 1a-b, MTF left unaltered body weight, serum glucose concentration and FDG clearance. By contrast, it almost halved estimated CMRGlu* and markedly decreased brain SUV (Fig. 1, Table 1).

Effect of metformin on brain glucose consumption and FDG uptake. a-b) CMRglu* and SUV maps in a control (green border) and ‘treated’ mouse (red border). c) Average time-course of ‘ex-vivo’ radioactivity expressed as fraction of the dose in brains harvested from control and MTF mice. d) Merged image of luxol staining and autoradiography of a representative brain. e) Fractional uptake of glucose (solid line) and FDG (dashed line) in control and MTF samples evaluated during the ex-vivo experiments. f) Experimental LC values as derived from e) (circles and solid line) as opposed to LC index measured according to HK function (squares and dashed line)
Ex vivo brain time activity curves showed the expected progressive increase in all experiments (Fig. 1c). Similarly, autoradiography documented, in all cases, a selective tracer uptake in the grey matter (Fig. 1d) confirming the viability of all studied brains for the experiment duration [16].
MTF decreased fractional FDG uptake decreased by almost 65% (from 7.3 ± 2.4% to 2.6 ± 1.2%, in control and ‘treated’ brains, respectively, p < 0.01) (Fig. 1e). However, fractional glucose removal from the incubation medium showed the opposite behaviour: it was 8 ± 2% under control conditions and more than doubled under MTF to 17 ± 3%, (p < 0.01) (Fig. 1e).
Thus, the MTF-induced decrease in CMRGlu* persisted ex vivo, occurred despite a marked increase in CMRGlu and obviously resulted in a marked reduction of LC that fell from the control value of 0.78 ± 0.09 to 0.17 ± 0.07 (p < 0.001) in treated brains (Fig. 1f).
Expression levels (Fig. 2a) and protein availabilities (Suppl Fig. 1) of most represented GLUTs [25] were not significantly affected by MTF that also did not modify gene expression of the sodium-glucose symporter (SGLT-1) (data not shown). Accordingly, MTF did not modify the hexose transport system represented in the whole brain lysate, suggesting a limited drug effect on hexose transport mechanisms across the blood-brain barrier. A similar consideration applies to \( \frac{1}{\phi } \): both mRNA levels (Fig. 2b) of the two isoforms (alpha and beta) [26] and total abundance of G6Pase (Suppl Fig. 1) did not change under MTF. More importantly, the drug did not affect overall enzyme activity nor its relative affinities for G6P and 2DG6P as FDG analogue (Fig. 2c).

Analysis of LC theoretical determinants. mRNAs levels in lysates of brains harvested from control and treated mice encoding for GLUTS (a) and G6Pase isoforms alpha and beta (b). c) G6Pase activity in the presence of G6P (solid columns) and 2DG6P (dashed columns). d) Real-time PCR of the four HK isoforms. e-f) Km and Vmax values of lysate HK activity in the presence of glucose (solid columns) or 2DG (dashed columns)
A similar invariance was also observed for HK. In agreement with previous literature [27], real-time PCR showed an almost exclusive HK I gene expression, with trace amounts of the other isoforms in both untreated and MTF brains (Fig. 2d). At the functional evaluation, the slight increase induced by MTF on Vmax was largely counterbalanced by the marked decrease in \( {K}_m^{\ast } \) (Fig. 2e-f) that even increased the product \( \frac{K_m}{K_m^{\ast }}\times \frac{V_{max}^{\ast }}{V_{max}} \).
We thus concluded the opposite effect of MTF on CMRGlu* and CMRGlu (Fig. 1f) cannot be explained by the classical model assuming cell accumulation of ‘the terminal metabolites’ FDG6P and 2DG6P. The increase in CMRGlu was largely explained by the MTF-induced inhibition of oxidative phosphorylation (OXPHOS), since complex I activity of brain lysates decreased from the control value 0.6 ± 0.05 to 0.4 ± 0.04 U × mg of proteins−1 in treated brains (p < 0.05, Fig. 3a). The subsequent acceleration of the glycolytic flux was documented by the increase in both PFK activity and lactate release (Fig. 3b-c). By contrast, periodic acid Schiff staining demonstrated that glycogen stores were scarcely represented in explanted brain cortex and remained constant after one-week of treatment (Fig. 3d). Finally, mRNA levels, catalytic activity (Fig. 3e-f) and protein abundance (Suppl Fig. 1) of G6PD remained unchanged under MTF. Thus, MTF effect on glycolysis, glycogen synthesis and PPP did not provide any possible insight into the mechanisms underlying the selective reduction in FDG uptake.

MTF effect on the determinants of G6P fate in brain. a-c) Complex I activity (a), PFK activity (b) and lactate release (c) in brain lysates of control (green) and treated (red) mice. d) Adjacent sections from brains of one control and one MTF mice stained with HE (left) and PAS for glycogen content (right). e-f) mRNA levels and enzymatic activity of G6PD (e-f) and of H6PD (g-h)
By contrast, the selective reduction in CMRGlu* was coherent with the expected response of H6PD: although MTF did not affect enzyme mRNA (Fig. 3g) and protein levels in brain lysates (Suppl Fig. 1), it substantially abolished H6PD catalytic activity. In fact, capability of cell lysates to dehydrogenate 2DG6P in the presence of NADP fell from the control value 3.30 ± 0.94 down to 0.54 ± 0.05 mU x mg of proteins (p < 0.01) (Fig. 3h).
Response of neuron and astrocyte metabolism to metformin
The opposite effect of MTF on CMRGlu* and CMRGlu and the adherence of the former to H6PD activity is actually disconcerting, since it challenges the undisputed consensus regarding the link between glycolytic flux and FDG uptake. However, this disagreement might reflect the heterogeneous nature of brain populating cells whose differences in metabolic pattern and sensitivity to MTF might eventually lead to an evident LC fall, despite an apparent invariance of its determinants in the whole brain lysates.
To test this hypothesis, we evaluated the metabolic response to 24 h MTF (10 mM) exposure of primary neurons and astrocytes, cultured according to our standard procedure. Control CMRGlu was almost 5-fold higher in astrocytes than in neurons (Fig. 4a) with an even more evident difference in glycolytic flux, as testified by almost 8-fold lactate release ( Fig. 4b). MTF increased CMRGlu, lactate release and PFK activity in both cell types (Fig. 4a-b). However, this response was particularly evident in neurons in agreement with their higher respiratory rate (Fig. 4d). Similarly, H6PD activity was almost 2-fold higher in neurons than in astrocytes under control conditions. MTF profoundly and selectively impaired this enzymatic function in neurons whose capability to dehydrogenate 2DG6P fell to levels even lower than those of control astrocytes (Fig. 4e). Intriguingly, cytosolic PPP showed an opposite pattern: G6PD catalytic function was markedly higher in astrocytes than in neurons under control conditions and did not respond to MTF in either cell type (Fig. 4f).

MTF effect on neurons and astrocytes. Glucose consumption (a) and lactate release (b) in control (solid column) and MTF-treated (for 24 h, dashed column) cultured astrocytes (orange) and neurons (blue). Activities of PFK (c), complex I (d), H6PD (e) and G6PD (f) for the two cell types in control and MTF conditions. Average intensity of ER (g) and 2-NBDG (h) signals as detected by confocal microscopy in control and MTF-treated astrocytes (orange) and neurons (blue). i) Costes analysis of signal colocalization expressed as percent of 2NBDG-containing voxels that colocalize with ER-positive voxels, for the two cell types in the two conditions
Handling of non-degradable hexose analogues in cultured neurons and astrocytes
To evaluate the cell compartmentalization of glucose-analogues uptake, we shifted toward the use of confocal microscopy and the fluorescent 2NBDG, whose uptake mechanism is thought to reproduce the kinetic model conventionally attributed to 2DG and FDG [28].
Intensity of ER signal was similar in both cell types and was not altered by MTF (Fig. 4g). Despite the large difference in glucose consumption, 2NBDG fluorescence was similar in neurons and astrocytes (Fig. 4h) under control conditions. However, MTF significantly impaired the uptake of the fluorescent 2DG analogue in neurons, as opposed to a significant increase observed in astrocytes (Fig. 4h). Interestingly, the response of tracer uptake to MTF was paralleled by an evident drug effect on signal location: MTF markedly and selectively decreased ER accumulation of 2NBDG in neurons. By contrast and coherently with the absent response of H6PD activity, it did not affect tracer distribution in astrocytes (Figs. 4i and 5).

Imaging of ER localization of 2NBDG in neurons and astrocytes. Representative images of ER (red), 2NBDG (green) and merged (white) signals in neurons (a) and astrocytes (b). Blue color reports the DAPI signal
Oxidative degradation of glucose and 2DG
This imaging analysis thus confirmed that H6PD activity is needed to preserve the direct relationship between FDG uptake and glucose consumption and documented that this same catalytic function configures the ER as the preferential accumulation site of its analogue 2NBDG. Together with the scarce substrate selectivity of this enzyme, these data thus suggest that ER metabolism might contribute to a further degradation of FDG6P and/or 2DG6P through an, at least partially, oxidative metabolism.
To verify this hypothesis, we estimated the oxygen consumption rate (OCR) and its response to MTF in isolated intact neurons and astrocytes incubated with glucose (20 mM) or 2DG (20 mM) (Fig. 6a-h). In agreement with the higher lactate/glucose ratio and with the lower mitochondrial complex I function, glucose-triggered OCR was markedly lower in astrocytes than in neurons under control conditions, while this difference disappeared under MTF. Unexpectedly, shifting available substrate from glucose to 2DG only slightly and not significantly affected OCR regardless of the cell type or treatment condition (Fig. 6i-j). Accordingly, this analysis confirmed that 2DG is indeed at least partially oxidized. This metabolic fate is well represented and contributes to preserving high FDG uptake, despite markedly lower glucose consumption in neurons with respect to astrocytes.

Respiration induced by 2DG in astrocytes and neurons. Original tracing of oxygen tension in incubation medium during the exposure of astrocytes (orange) and neurons (blue) to glucose (a and c) or 2DG (b and d) under control conditions. e-h: same experimental conditions for astrocytes and neurons incubated 24 h with MTF. At the end of the measurements, absent response to ADP confirmed cell integrity. Average OCR of astrocytes (i) and neurons (j) cultured in control (solid) and MTF (dashed) conditions and challenged with glucose or 2DG
Discussion
The main finding of our study is that brain uptake of the 2DG analogues, FDG and 2NBDG, is largely independent of cytosolic glucose consumption and reflects the activation of a specific metabolic pathway triggered by H6PD within the ER. This conclusion agrees with our previous experiments in cancer [9] and is based on the following observations: i) brain lysates contained measurable H6PD activity, as demonstrated by their capability to produce NADPH in the presence of 2DG6P; ii) the steep fall in the CMRGlu*/CMRGlu ratio, induced by MTF, was coherent with the drug-induced inhibition of H6PD activity, while it was not explained by any measurable change in the acknowledged determinants of LC (transmembrane transport, G6Pase activity and hexose phosphorylation kinetics); iii) the markedly higher H6PD activity empowered cultured neurons with a similar uptake of 2NBDG despite a five-fold lower glucose consumption and even lower rates of glycolytic flux with respect to astrocytes; iv) ER localization of 2NBDG was well represented in neurons, while it was impaired when enzyme function was less expressed (in astrocytes) or hampered (by MTF), and, finally, v) 2DG was at least partially oxidized both in neurons and astrocytes, challenging the universally assumed ‘dead-end’ nature of its phosphorylated form.
The conventional 3-compartment model (Fig. 7a) considers that the FDG entry-gate to the non-exchangeable pool is the HK-catalyzed phosphorylation (k3). The present data move this gate to a downstream reaction (or sequence of reactions) triggered by H6PD within the ER, clarifying the still unexplained brain loss of 2DG6P/FDG6P that has been systematically documented since the introduction of the 2DG method [1, 19, 29]. This finding has been generically attributed to G6Pase-catalyzed hydrolysis; however, this assumption conflicts with the well-documented enzyme sequestration in the ER lumen [30]. Since electrically-charged hexoses cannot cross the ER membrane, they need unidirectional transport mechanisms to access to the hydrolytic enzyme, while back-diffusion of dephosphorylated counterparts is warranted by the large GLUT abundance of the ER membrane [25]. In agreement with this concept, FDG radioactivity release has been found to reflect the expression of the ATP-dependent G6P transporter more than G6Pase activity [31]. The proposed model would thus configure a true accumulation kinetics, considering FDG6P/2DG6P as a reversible compartment that can undergo the competitive action of H6PD and G6Pase for dehydrogenation (and storage) or de-phosphorylation (and release), respectively. In the presence of a high H6PD activity, FDG6P is processed towards substrates retained within the ER (Fig. 7b). By contrast, when H6PD function is scarcely expressed (e.g. in astrocytes) or inhibited (by MTF) (Fig. 7c), G6Pase activity becomes prevalent, eventually decreasing tracer retention. This configuration agrees with previous evidence reporting a further processing of FDG6P [5,6,7,8,9] and is further corroborated by the evident OCR triggered by 2DG and its sensitivity to the combined inhibition of H6PD and Complex I by MTF. Obviously, this observation does not configure the 2DG-derived metabolites as true respiratory substrates: to this purpose the coupling between OCR and ATP synthesis should have been evaluated. This task was not feasible in our experimental model using intact cells. However, the evident OCR documents ‘per se’ that 2DG6P catabolism occurs, regardless of its coupling with ATP synthesis or of the generation of reactive oxygen species, and contributes to the ultimate radioactivity retention.

The possible models explaining FDG uptake. Panel a displays the conventional 3-compartment models assuming the accumulation of the terminal product FDG6P in the cytosol. Panel b displays the proposed model in neurons in which the thick arrow indicates a high H6PD activity and its capability of storing obtained intermediates for later degradation. In Panel c, the low H6PD activity results in a measurable FDG6P de-phosphorylation by the competing G6Pase eventually increasing tracer loss
This paradigm shift sheds a new light on the highly animated discussion about the different rates and scopes of glucose consumption in neurons and astrocytes [32, 33]. Under control conditions, these two cell types showed similar levels of 2NBDG uptake, despite an almost 5-fold higher glucose consumption (and an even higher difference in glycolytic flux) in astrocytes with respect to neurons. This mismatch was explained by the different rates of H6PD activity in the two cell types and the preferential OXPHOS nature of neuronal glucose usage. In agreement with these features, H6PD-inhibition by MTF severely impaired the relationship between 2NBDG uptake and glucose disposal in neurons, while it was markedly less effective in astrocytes. Accordingly, these data suggest that estimation of 2DG/FDG uptake should be carefully compared with direct measurements of glucose consumption in the two different cell types.
On methodological grounds, inhibition of H6PD function was the unique possible explanation for the steep fall in CMRGlu/CMRGlu* ratio – and thus in LC – observed in MTF brains. Actually, estimation of brain LC provided largely different results, ranging from 0.52 [18] up to 1.24 [21]. Although a number of methodological limitations and peculiarities have been claimed, this disappointing variability has not been fully explicated and, more importantly, no conclusive explanation has been proposed for its dependence upon experimental conditions. Indeed, LC has been found to rapidly increase with decreasing glucose availability both in vitro (from 0.7 to 1.22) [20] and in vivo (from 0.41 to 1.24) [21]. Similarly, Blomqvist et al. found a strict and direct link between regional neuronal activation and corresponding LC value [22]. These findings intrinsically suggest that the difference between CMRGlu* and CMRGlu cannot be explained by the conventional approach, assuming differences in the affinities of GLUTs, HK or G6Pase for the two hexoses. By contrast, they nicely agree with the dependence of FDG uptake upon the activity of the oxidative PPP phase, triggered by H6PD within the ER, and suggest that neuronal activation might result in a preferential glucose oxidation and FDG uptake (through H6PD function).
In conclusion, the present study documents that brain uptake of 2DG, FDG and 2NBDG cannot be considered the accumulation of ‘inert analogues of phosphorylated glucose’ and thus does not provide accurate estimations of local glycolytic flux. It rather selectively reflects the rate of a still undefined ER metabolic machinery triggered by the omnivore enzyme H6PD, which is able to degrade phosphorylated and free hexoses. The disconcerting nature of this conclusion needs further investigation to define presence, rate, significance and control mechanisms of this unknown metabolism, at least ‘geographically’ independent from the two cytosolic pathways, glycolysis and PPP, commonly assumed responsible for glucose degradation. Should this concept be extended to the regional agreement between signal processing and CMRGlu, it would provide a new insight into the cellular basis of metabolic imaging of brain function and would possibly permit the development of tracers selectively targeting the ER activation as a potential contribution to brain disorders.
References
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, et al. The 14C-deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916.
Varrone A, Asenbaum S, Vander-Borght T, Booij J, Nobili F, Någren K, et al. EANM procedure guidelines for PET brain imaging using [18F]-FDG. Eur J Nucl Med Mol Imaging. 2009;36:2103–10.
Sols A. Substrate specificity of brain hexokinase. J Biol Chem. 1954;210:581–95.
Bachelard HS. Specificity and kinetic properties of monosaccharide uptake into Guinea pig cerebral cortex in vitro. J Neurochem. 1971;18:213–22.
Dienel GA, Cruz NF, Sokoloff L, Driscoll BF. Determination of glucose utilization rates in cultured astrocytes and neurons with [14C] deoxyglucose: progress, pitfalls, and discovery of intracellular glucose compartmentation. Neurochem Res. 2017;42:50–63.
Kanazawa Y, Yamane H, Shinohara S, Kuribayashi S, Momozono Y, Yamato Y, et al. 2-Deoxy-2-Fluoro-D-glucose as a functional probe for NMR: the unique metabolism beyond its 6-phosphate. J Neurochem. 1996;66:2113–20.
Dienel GA, Cruz NF. Synthesis of deoxyglucose-1-phosphate, deoxyglucose1,6-biphosphate, and other metabolites of 2-deoxy-D-[14C]glucose in rat brain in vivo: influence of time and tissue glucose level. J Neurochem. 1993;60:2217–31.
Southworth R, Parry CR, Parkes HG, Medina RA, Garlick PB. Tissue-specific differences in 2-fluoro-2-deoxyglucose metabolism beyond FDG-6-P: a 19 F NMR spectroscopy study in the rat. NMR Biomed. 2003;16(8):494–502.
Marini C, Ravera S, Buschiazzo A, Bianchi G, Orengo AM, Bruno S, et al. Discovery of a novel glucose metabolism in cancer: the role of endoplasmic reticulum beyond glycolysis and pentose phosphate shunt. Sci Rep. 2016;6:25092.
Senesi S, Csala M, Marcolongo P, Fulceri R, Mandl J, Banhegyi G, et al. Hexose-6-phosphate dehydrogenase in the endoplasmic reticulum. Biol Chem. 2010;391:1–8.
Kulkarnit P, Hodgson E. Mouse liver microsomal hexose-6-phosphate dehydrogenase. Biochem Pharmacol. 1982;31:1131–7.
Moreira PI. Metformin in the diabetic brain: friend or foe? Ann Transl Med. 2014;2:2–4.
Correia S, Carvalho C, Santos MS, Proença T, Nunes E, Duarte AI, et al. Metformin protects the brain against the oxidative imbalance promoted by type 2 diabetes. Med Chem. 2008;4:358–64.
Blumrich EM, Dringen R. Metformin accelerates glycolytic lactate production in cultured primary cerebellar granule neurons. Neurochem Res. 2017:1–12. https://doi.org/10.1007/s11064-017-2346-1.
Westhaus A, Maria E, Dringen R. The antidiabetic drug metformin stimulates glycolytic lactate production in cultured primary rat astrocytes. Neurochem Res. 2015;123:1–16.
Buschiazzo A, Cossu V, Bauckneht M, Orengo A, Piccioli P, Emionite L, et al. Effect of starvation on brain glucose metabolism and 18F-2-fluoro-2- deoxyglucose uptake: an experimental in-vivo and ex-vivo study. EJNMMI Res. 2018. https://doi.org/10.1186/s13550-018-0398-0.
Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1–7.
Reivich M, Kuhl D, Wolf A, Greenberg J, Phelps M, Ido T, et al. The 18F-fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ Res. 1979;44(1):127–37.
Graham MM, Muzi M, Spence AM, O'Sullivan F, Lewellen TK, Link JM, et al. The FDG lumped constant in normal human brain. J Nucl Med. 2002;43:1157–67.
Noll T, Mühlensiepen H, Engels R, Hamacher K, Papaspyrou M, Langen KJ, et al. A cell-culture reactor for the on-line evaluation of radiopharmaceuticals: evaluation of the lumped constant of FDG in human glioma cells. J Nucl Med. 2000;41:556–64.
Suda S, Shinohara M, Miyaoka M, Lucignani G, Kennedy C, Sokoloff L. The lumped constant of the deoxyglucose method in hypoglycemia: effects of moderate hypoglycemia on local cerebral glucose utilization in the rat. J Cereb Blood Flow Metab. 1990;10:499–509.
Blomqvist G, Seitz RJ, Sjögren I, Halldin C, Stone-Elander S, Widén L, et al. Regional cerebral oxidative and total glucose consumption during rest and activation studied with positron emission tomography. Acta Physiol Scand. 1994;151:29–43.
Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003.
Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. J Biol Chem. 1925;66:375–400.
Cura AJ, Carruthers A. Role of monosaccharide transport proteins in carbohydrate assimilation , distribution, metabolism , and homeostasis. Compr Physiol. 2012;2:863–914.
Guionie O, Clottes E, Stafford K, Burchell A. Identification and characterisation of a new human glucose-6-phosphatase isoform. FEBS Lett. 2003;551:159–64.
Muzi M, Freeman SD, Burrows RC, Wiseman RW, Link JM, Krohn KA, et al. Kinetic characterization of hexokinase isoenzymes from glioma cells: implications for FDG imaging of human brain tumors. Nucl Med Biol. 2001;28:107–16.
Zou C, Wang Y, Shen Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J Biochem Biophys Methods. 2005;64:207–15.
Sokoloff L. Relation between physiological function and energy metabolism in the central nervous system. J Neurochem. 1977;29:13–26.
Van Schaftingen E, Gerin I. The glucose-6-phosphatase system. Biochem J. 2002;532:513–32.
Caracó C, Aloj L, Chen LY, Chou JY, Eckelman WC. Cellular release of [18F]2-Fluoro-2-deoxyglucose as a function of the glucose-6-phosphatase enzyme system. J Biol Chem. 2000;275:18489–94.
Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Neurobiology. 1994;91:10625–9.
Dienel GA. Review lack of appropriate stoichiometry: strong evidence against an energetically important astrocyte–neuron lactate shuttle in brain. J Neurosci Res. 2017;95:2103–25.
Acknowledgements
The authors are indebted with Proff. Antonio De Flora, Alessandro Morelli and Alberto Pupi for the enthusiastic support and criticisms.
Funding
This study was funded by the program “Ricerca Corrente,” line “Guest-Cancer Interactions,” by Compagnia di San Paolo (project ID Prot.: 2015.AAI4110.U4917).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
All procedures involving animals were performed in respect of the current National and International regulations and were reviewed and approved by the Licensing and Animal Welfare Body of the IRCCS Ospedale Policlinico San Martino, Genoa, Italy and by the Italian Ministry of Health.
Conflict of interests
No author has any conflict of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cossu, V., Marini, C., Piccioli, P. et al. Obligatory role of endoplasmic reticulum in brain FDG uptake. Eur J Nucl Med Mol Imaging 46, 1184–1196 (2019). https://doi.org/10.1007/s00259-018-4254-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-018-4254-2