
Abstract
Purpose
Two allosteric modulators of the group I metabotropic glutamate receptors (mGluR1 and mGluR5) were evaluated as positron emission tomography (PET) radioligands for mGluR1.
Methods
LY2428703, a full mGluR1 antagonist (IC50 8.9 nM) and partial mGluR5 antagonist (IC50 118 nM), and LSN2606428, a full mGluR1 and mGluR5 antagonist (IC50 35.3 nM and 10.2 nM, respectively) were successfully labeled with 11C and evaluated as radioligands for mGluR1. The pharmacology of LY2428703 was comprehensively assessed in vitro and in vivo, and its biodistribution was investigated by liquid chromatography-mass spectrometry/mass spectrometry, and by PET imaging in the rat. In contrast, LSN2606428 was only evaluated in vitro; further evaluation was stopped due to its unfavorable pharmacological properties and binding affinity.
Results
11C-LY2428703 showed promising characteristics, including: (1) high potency for binding to human mGluR1 (IC50 8.9 nM) with no significant affinity for other human mGlu receptors (mGluR2 through mGluR8); (2) binding to brain displaceable by administration of an mGluR1 antagonist; (3) only one major radiometabolite in both plasma and brain, with a negligible brain concentration (with 3.5 % of the total radioactivity in cerebellum) and no receptor affinity; (4) a large specific and displaceable signal in the mGluR1-rich cerebellum with no significant in vivo affinity for mGluR5, as shown by PET studies in rats; and (5) lack of substrate behavior for efflux transporters at the blood–brain barrier, as shown by PET studies conducted in wild-type and knockout mice.
Conclusion
11C-LY2428703, a new PET radioligand for mGluR1 quantification, displayed promising characteristics both in vitro and in vivo in rodents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metabotropic glutamate receptors (mGluRs) are a family of G protein-coupled receptors classified into three groups based on their sequence homology, type of signal transduction pathway, and pharmacology [1]. Group I includes two receptors that are structurally very similar: mGluR1 and mGluR5. Both stimulate phospholipase C to hydrolyze phosphoinositide phospholipids and mobilize intracellular calcium [2]. Several studies have demonstrated that mGluR1 is involved in spatial and associative learning [3], as well as in regulating synaptic plasticity in the hippocampus and cerebellum [4]. mGluR1 has also been linked to the pathophysiology of several neurological and psychiatric disorders, such as Parkinson’s disease, Huntington’s disease, stroke, epilepsy, anxiety and stress disorders, drug and alcohol addiction, and pain (see [4] for a review). In vivo quantification with an appropriate radioligand would help clarify the role of mGluR1 in these diseases, which may lead to the development of new therapies.
While different imaging compounds for mGluR5 have been synthesized and tested in vivo in both animals and humans [5–7], a detailed study of mGluR1 has hitherto been hindered by the lack of high-affinity and selective ligands for this receptor subtype. A handful of potential mGluR1 ligands have been described in the literature [8–14] but, in vivo, only some of these have shown promising characteristics in animal models. Therefore, there is a considerable unmet need to develop radioligands capable of selectively imaging glutamate receptors in the living brain, so that the role of these receptors in health and neuropsychiatric disorders can be better elucidated. Such radioligands may themselves become useful for drug discovery and assessment of the efficacy of neuropsychiatric therapies and treatments.
We describe here the synthesis and evaluation of two potential candidate radioligands—11C-LY2428703 and 11C-LSN2606428—that act as antagonists for mGluR1. 11C-LY2428703 was extensively evaluated in vitro and in vivo using liquid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS) biodistribution studies and positron emission tomography (PET) imaging in rodents. These studies showed that 11C-LY2428703 has promising characteristics as a candidate radiotracer for mGluR1. 11C-LSN2606428 was abandoned after initial in vitro evaluation due to its less favorable pharmacological properties and binding affinity compared to 11C-LY2428703.
Materials and methods
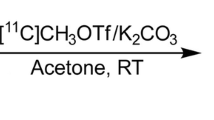
Synthesis of both ligands and the radiolabeling methods are described in the Supplementary material. The labeling schemes and chemical structures of 11C-LY2428703 and 11C-LSN2606428 are given in Fig. 1.

Reaction schemes and chemical structures of 11C-LSN2606428 (top) and 11C-LY2428703 (bottom)
In vitro experiments
The following in vitro evaluations were carried out:
-
1.
Measurement of logD7.4 values in octanol for both 11C ligands.
-
2.
Pharmacological characterization of LY2428703 and LSN2606428 by measuring the effects of each compound on glutamate-evoked mobilization of intracellular calcium from cells expressing the human mGlu1, mGlu2, mGlu3, mGlu4, mGlu5, mGlu7, and mGlu8 receptors. Agonist effects were quantified as the percentage stimulation induced by the compound alone relative to the maximal glutamate response. Antagonist effects were quantified by calculating the percentage inhibition of the EC90 glutamate response caused by the compound. Potentiation effects were quantified as the percentage increase in the presence of an EC10 response in glutamate relative to the ECmax response.
-
3.
Competition binding assays with 3H-LSN456066, a potent and selective radioligand for mGluR1 [15] to test the affinity of both ligands for the target across different species (rat, monkey, and human).
On the basis of its unfavorable pharmacological properties and binding affinity, further development of LSN2606428 as a human PET imaging agent was stopped. Therefore, the following in vitro analyses, as well as the subsequent in vivo analyses, were conducted only with the more promising LY2428703:
-
1.
Metabolic stability in liver microsomes across species (human, monkey, dog, rat, and mouse). Fast gradient elution LC-ESI/MS with column switching was used to estimate the percentage loss of a substrate following phase I metabolism in hepatic microsomes over a 30-minute incubation period.
-
2.
Assay for P-glycoprotein (P-gp) substrate identification using MDCK cells transfected with wild-type MDR1.
-
3.
Screen for brain exposure with a calibrated mouse brain uptake assay.
-
4.
Plasma and brain unbound fractions, measured by equilibrium dialysis.
-
5.
In vitro stability of labeled LY2428703 in rat whole blood and brain homogenate, using a radio high-performance liquid chromatography (HPLC) analysis.
A detailed description of each in vitro procedure is provided in the Supplementary material.
Ex vivo experiments
Animals
Adult male Sprague-Dawley rats (Harlan Laboratories, Indianapolis, IN) weighing approximately 230–300 g were housed two or three per cage in rooms using a 12-h light/dark cycle (lights on at 6 a.m.). Room temperature was maintained at 21 ± 3 °C. Rats had access to normal rat chow and water ad libitum until the beginning of the 4-h experimental protocol. Rats were permitted at least 2 days of acclimation to the housing environment prior to testing. All studies were performed in accordance with the National Research Council Guide under protocols approved by the Animal Care and Use Committee of Eli Lilly and Company.
The following ex vivo evaluations were carried out:
-
1.
Plasma and brain radiochromatographic profile, to determine whether 11C-LY2428703 could cross the blood–brain barrier.
-
2.
LC-MS/MS rat brain kinetic analysis with nonlabeled tracer.
-
3.
Pharmacological validation of LY2428703 as an mGluR1 tracer using JNJ16259685, a selective mGluR1 antagonist.
-
4.
mGluR1 and mGluR5 occupancy experiments to assess in vivo selectivity of LY2428703.
A detailed description of each ex vivo procedure, along with the methodology for tissue processing, LC-MS/MS analysis and receptor occupancy calculations, is provided in the Supplementary material.
In vivo experiments
PET imaging
Rats and mice were anesthetized with 1.5 % isoflurane in oxygen and were injected with a bolus of 11C-LY2428703 in a tail vein using an automated pump. PET brain imaging studies were carried out with a Focus 120 or Focus 220 animal scanner (Siemens Medical Solutions, Knoxville, TN). Serial dynamic image data acquisition was begun at the time of injection and was continued for 100 min with a frame schedule of 20 s × 6, 60 s × 5, 120 s × 4, 300 s × 5, 600 s × 5, and 1,200 s × 2. The resulting time–activity curves were plotted in terms of standardized uptake values (SUV). All studies were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Specific binding
PET imaging was used to determine in vivo specific binding. Four Sprague-Dawley rats were pretreated with one of two mGluR1 antagonists, either 10 mg/kg of LY2212157 or 3 mg/kg of LY2332084, 30 min prior to radiotracer injection. In addition, a displacement study was conducted by administering LY2332084 (3 mg/kg) to another rat 10 min after radiotracer injection. The brain time–activity curves, drawn over the cerebellum and the rest of the brain, were compared to those of ten rats at baseline. The average weight of all 17 rats was 337 ± 97 g, and the average injected activity was 38.7 ± 6.4 MBq (0.699 ± 0.316 nmol).
Cross-reactivity with mGluR5 receptors
PET imaging was also used to determine cross-reactivity with mGluR5 receptors. Two rats were pretreated with 5 mg/kg of MTEP (3-((2-methyl-4-thiazolyl)ethynyl)pyridine), an mGluR5 antagonist, 30 min before radioligand injection. The curves were compared to those of ten rats at baseline.
Permeability at the blood–brain barrier
PET imaging was used to assess whether 11C-LY2428703 is a substrate for efflux proteins at the blood–brain barrier using knockout mice. Four ABCB1 (P-gp) knockout mice (FVB.129P2-Abcb1a tm1Bor/Abcb1b tm1Bor N12), four ABCG2 (BCRP) knockout mice (FVB.129P2-Abcg2 tm1Ahs N7), and four ABCB1 + ABCG2 triple knockout mice (FVB.129P2-Abcb1a tm1Bor/Abcb1b tm1Bor/Abcg2 tm1Ahs N7) (Taconic Farm, Germantown, NY) were scanned after injection of 11C-LY2428703. The brain time–activity curves were compared to those obtained in five wild-type mice. The average weight of the mice was 28 ± 5.6 g, and the injected activity was 12.6 ± 5.0 MBq (0.336 ± 0.157 nmol).
Statistical analyses
Prism (GraphPad Software Inc., version 4.0, San Diego, CA) software was employed for calculations, curve fitting, and graphics. Sigmoidal dose–occupancy curve was calculated using a four-parameter (top, bottom, slope, and ED50) logistic fit. The relative ED50 values, the occupancy seen at a point on the curve half way between the calculated curve top and bottom, are reported.
Results
Radiochemistry
11C-LSN2606428 was prepared by treating the N-desmethyl analog with 11C-methyl triflate without added base. 11C-LY2428703 was prepared by treating the phenol LY2483097 with 11C-methyl triflate in the presence of added base. These radioligands were separated in high radiochemical purity; chemical purity and specific radioactivity were determined by reverse-phase HPLC and readily formulated for intravenous injection.
In vitro experiments
LogD7.4 measurements
The measured lipophilicity index, logD7.4, was 4.02 ± 0.09 (n = 6) for 11C-LY2428703, and 1.42 ± 0.14 (n = 6) for 11C-LSN2606428. The calculated logP values were 3.1 and 2.9, respectively.
Pharmacological characterization of LY2428703 and LSN2606428
Pharmacological profiling in human receptors showed that LY2428703 had a higher affinity for the target than LSN2606428 (IC50 8.9 and 35.3 nM, respectively). Moreover, LSN2606428 also showed significant cross-affinity for mGluR5 (IC50 10.2 nM). In contrast, LY2428703 had a more than tenfold greater affinity for mGluR1 than for mGluR5 (IC50 8.9 and 118 nM, respectively). Neither compound had significant affinity for other mGluRs or off-target cross-reactivity. No agonist effects or postpotentiation effects were noted (Tables 1 and 2).
Competition binding assay with 3H-LSN456066
In all the species studied, the competition binding assay demonstrated that the affinity of LY2428703 was much higher than the affinity of LSN2606428 (Table 3), especially in humans, where the affinity was about 20-fold higher. In light of the in vitro results of the pharmacological characterization of the tracers and the competition binding assay, only LY2428703 was used for further analyses.
Metabolic stability across species
The hepatic microsomes study showed that the metabolic stability of LY2428703 was greater in humans than in other species. After 30 min, only 37.1 ± 29.9 % of the compound was metabolized in humans (n = 2), compared to at least 65 % in other species (mouse, n = 3, 79.5 ± 14.7 %; rat, n = 2, 91.1 ± 3.4 %; dog, n = 3, 65.0 ± 15.5 %; monkey, n = 2, 90.1 ± 3.1 %).
Assay for P-gp substrate identification
The absence of net asymmetrical flux (e.g., the B-A/A-B flux ratio was about 1.0) across monolayers of polarized MDCK cells overexpressing human P-gp at the apical surface indicated that LY2428703 was not a P-gp substrate. LY2428703 had rapid passive permeability, with a permeability coefficient of 620 ± 30 nm/s (n = 8), which is approximately equal to the aqueous boundary layer-limited permeability in this model system. Paracellular leakage for mannitol was about 7 nm/s. Mass balance recovery for LY2428703 was 108 ± 10 % (n = 8).
Screen for brain exposure
The calibrated mouse brain uptake assay (cMBUA) was used to screen compounds. Blood–brain barrier penetration of LY2428703 was fast, with a permeability coefficient of 190 nm/s, which is equal to cerebral blood flow as judged by reference compounds such as testosterone and tritiated water [16]. The percentage of the dose delivered to the brain was 1.2–1.6 % over the tenfold range of doses administered, which is similar to brain levels of tritiated water at 1.9 ± 0.2 % of the dose, and within the range of cardiac output (1.2–2.6 %) reported for the rat [16, 17]. Clearance from plasma and brain, represented as the percentage loss in concentration from 5 to 60 min, was similar and >95 %.
Measuring the unbound fraction
Using equilibrium dialysis, unbound fractions in plasma (rat) and brain homogenate (dog) were measured as 0.027 and 0.014, respectively. Assuming that protein binding is similar in mouse, the Cbrain,u/Cplasma,u ratio, or a K p,uu value [18], was calculated as 0.7 to 1.0 for 5 and 60 min from the cMBUA data.
In vitro stability of 11C-LY2428703 in rat whole-blood and brain homogenates
The stability of 11C-LY2428703 was verified in rat whole-blood and brain homogenates over 1 h at 37 °C. Enough radioactivity was added to detect a radiochemical impurity as low as 0.5 %. Radiochemical purity was 100 %. Within these conditions, no biotransformation products were detected.
Ex vivo experiments
Plasma and brain radiochromatographic profile
Plasma samples were obtained from rats 30 min after radioligand injection. One major radiometabolite (17.2 %), eluted in the void volume of the column, was detected with a minor one (1.3 %) that was more lipophilic than the first radiometabolite but less lipophilic than the parent. The parent radioligand was 75 % of the plasma radioactivity. The chromatogram from the UV absorbance detector confirmed the identity of 11C-LY2428703 because the extracted sample was spiked with reference compound.
Radiometabolite concentrations in brain regions were less than plasma concentrations. The rat brain and cerebellar radioactivity comprised mainly parent radioactivity and a radiometabolite less lipophilic than the parent. The relative composition differed between the two brain regions. The brain was composed of 82.7 % parent radioligand and 17.2 % radiometabolite; cerebellar activity was composed of 96.2 % parent radioligand and 3.7 % radiometabolite. The %SUV concentration of 11C-LY2428703 in brain, cerebellum, plasma, and urine was 49, 156, 122, and 2.7, respectively. The %SUV concentration of the major radiometabolite in brain, cerebellum, plasma, and urine was 10.2, 6.1, 38.5, and 161, respectively. For the parent, the cerebellum/brain concentration ratio was 3.2, the cerebellum/plasma ratio was 1.3, and the brain/plasma ratio was <1.0. The same regional ratios of the radiometabolite were always <1.0.
Radioactivity analysis of the brain and cerebellar tissues without blood yielded very similar results. Using the mean of two separate measurements, cerebellar activity was composed of 95.8 % (169 %SUV) parent radioligand and 3.3 % (5.6 %SUV) radiometabolite. Brain activity was composed of 82.5 % (48 %SUV) parent radioligand and 16.5 % (9.5 %SUV) radiometabolite.
LC-MS/MS rat brain kinetic analysis with nonlabeled tracer
LY2428703 differentially distributed towards the target-rich cerebellum relative to the frontal cortex at all time points, indicating specific binding at all time points. The time-course study showed rapid uptake of the tracer and washout in a time frame amenable for PET ligand 11C imaging, given its half-life of 20.4 min. The 10 μg/kg dose of LY2428703 reached 330 % SUV in rat cerebellum at 15 min, the earliest time point measured (Fig. 2).

Time-course of cerebellar (black circles) and cortical (white circles) tracer levels of LY2428703. The differential uptake between the target-rich cerebellum and the frontal cortex indicates specific binding at all time points
The intravenous administration of the 3 μg/kg dose of LY2428703 with a survival interval of 40 min was chosen for the dose–response study with the blocker JNJ16259685, because it afforded the largest signal to noise ratio of total (cerebellum) to nonspecific (frontal cortex) binding without compromising detection and quantitation of LY2428703 in rat brain tissue by LC-MS/MS. The tracer dose was selected to be as low as possible, in order to mimic the PET imaging situation. The time between tracer delivery and the ex vivo evaluation was as long as possible to increase the signal without compromising LC-MS/MS detection.
Pharmacological validation of LY2428703 as an mGluR1 tracer
The selective mGluR1 antagonist JNJ16259685 dose-dependently reduced the level of specific binding of the nonlabeled tracer LY2428703. This resulted in an in vivo mGluR1 ED50 of 1.4 ± 0.1 mg/kg following intravenous administration, demonstrating that JNJ16259685 effectively blocked the specific binding of LY2428703 when the latter was used as a tracer.
mGluR1 and mGluR5 occupancy experiments to assess in vivo selectivity of LY2428703
To evaluate the in vivo selectivity of LY2428703, rats were pretreated with a selective mGluR1 antagonist, a selective mGluR5 antagonist, or vehicle. LY2428703 dose-dependently occupied rat mGluR1s following oral administration, resulting in dose decreases in nonlabeled JNJ16259685. Conversely, despite modest mGluR5 partial antagonist functional potency, LY2428703 at doses up to 60 mg/kg (about 200 μmol/kg) did not appear to show any significant occupancy of mGluR5 under the same treatment paradigm and with the same animals; that is, the same drug exposure and the modest potency in vitro did not translate into in vivo mGluR5 occupancy (Fig. 3).

Calculated LY2428703 occupancy of mGluR1 and mGluR5. While mGluR1 occupancy increases with increasing doses of LY2428703, little to no mGluR5 occupancy was found up to doses of 60 mg/kg
PET in vivo experiments
Specific binding
11C-LY2428703 accumulated preferentially in rat cerebellum, and only faint uptake was seen in the rest of the brain (Fig. 4). This uptake disappeared in the blocked scans, suggesting a large specific and displaceable signal (Fig. 5).

11C-LY2428703 PET studies in a rat at baseline (a) and after blocking using LY2212157 (10 mg/kg) (b). The strongest signal at baseline is in the cerebellum, which has disappeared after blockade (arrows). The rest of the brain (circles) shows a faint uptake at baseline, which has disappeared after blockade. The residual activity is nonspecific binding. Note that the two areas of high uptake visible at both baseline and during the blocked study in the anterior part of the head represent nonspecific uptake in the orbital regions

Time–activity curves for 11C-LY2428703 in rat forebrain and cerebellum. a Baseline (black symbols; n = 10) and blocked (LY2212157, 10 mg/kg; white symbols; n = 4) curves for the cerebellum (black circles) and forebrain (black squares). b Displacement study in one rat. The displacer (LY2212157, 10 mg/kg) was administered 15 min after the radiotracer injection, which led to a rapid decrease in cerebellar activity (white circles) to the level of forebrain activity (black circles)
Cross-reactivity with mGluR5 receptors
Echoing the results of the aforementioned in vitro receptor occupancy studies, in vivo PET studies showed that 11C-LY2428703 had negligible affinity for mGluR5 in vivo (Fig. 6).

Time–activity curves for 11C-LY2428703 in rat forebrain (squares) and cerebellum (circles) without (baseline; black symbols; n = 10) and with (white symbols; n = 2) mGluR5 antagonist (MTEP, 5 mg/kg) pretreatment. Forebrain uptake was not affected by pretreatment with an mGluR5 antagonist
Permeability at the blood–brain barrier
In line with the results obtained via MDCK cell assay, comparison of the brain time–activity curves obtained in wild-type and knockout mice showed that 11C-LY2428703 was not a substrate for either ABCB1 or ABCG2 transporter at the blood–brain barrier in mice (Fig. 7).

Time–activity curves in the cerebellum (circles) and forebrain (squares) of wild-type mice (black symbols; n = 5) and knockout mice (white symbols) for P-gp (a, n = 4) and BCRP (b, n = 4). Very similar results were obtained with triple knockout mice (n = 4) (data not shown)
Discussion
This study investigated the in vitro, ex vivo, and in vivo characteristics of 11C-LY2428703, and showed that this compound is an excellent potential PET ligand for mGluR1s.
First, we assessed in vitro the affinity and selectivity of two different candidate tracers: LY2428703 and LSN2606428. LY2428703 was found to have a high affinity for the target (IC50 8.9 nM) and very small cross-reactivity with mGluR5s (IC50 118 nM). In contrast, LSN2606428 not only had a lower affinity for mGlu1Rs (IC50 35.3 nM), but also displayed a significant cross-affinity for mGluR5 (IC50 10.2 nM). Moreover, although both tracers displayed a high affinity in rodents, LY2428703 had a significantly higher affinity than LSN2606428 in monkeys (2.1 and 31.4 nM, respectively), and even higher affinity in humans (1.4 in females and 2.7 in males for LY2428703, >50 nM for LSN2606428). As a result, only LY2428703 was used in further analyses.
LY2428703 has favorable physical/chemical attributes for brain exposure (calculated logP 3.1, one hydrogen bond donor, four hydrogen bond acceptors, polar surface area of 51 Å2) that are similar to the median values (calculated logP 3.0, one hydrogen bond donor, four hydrogen bond acceptors, polar surface area of 49 Å2) of 20 PET ligands drawn from published studies and tested to date for various brain targets, both receptors and enzymes (T.J. Raub, unpublished data). Consequently, the measured activity of LY2428703 was similar to the median values for the PET ligand set tested. The in vitro passive permeability coefficient was 600 nm/s compared to the median of 620 nm/s, and absence of P-gp efflux was the norm. These data and a K p,uu of unity show that LY2428703 passively crossed the blood–brain barrier. The 1.6 ± 0.2 % of the dose distributed to the brain was 84 % of cardiac output, and was less than the median value of 2.7 % of the dose for the PET ligand test set, but within the measured range of 0.5 to 6.7 % (see reference [16] and T.J. Raub, unpublished data). Another desirable behavior for a prospective PET ligand is rapid and reversible equilibration, such that nonspecific levels in the brain decrease in parallel to systemic clearance. The median losses from plasma and brain for the PET ligand test set were 90 % and 94 %, respectively, and >95 % LY2428703 was lost from both compartments, confirming that it displayed this desired behavior.
Another favorable characteristic of LY2428703 was its metabolic stability, which was greater in humans than in other species. In fact, 30 min after injection, only about 37 % of the compound was metabolized in human hepatic microsomes compared to at least 65 % in mice, rats, dogs, and monkeys. Moreover, the radioligand was stable in whole blood in vitro, which suggests that its concentrations in blood are amenable to a reliable in vivo measurement when an arterial input function is needed.
In order to assess whether radiometabolites were present and able to cross the blood–brain barrier, we measured the plasma, brain, and cerebellum radio HPLC radiochromatographic profiles of 11C-LY2428703. Accumulation of radiometabolites in the brain would prevent the stable measurement of the distribution volume of the parent through kinetic modeling. HPLC analysis of 11C-LY2428703 in rat plasma and brain showed that one relatively polar radiometabolite was produced in plasma. However, the penetration of this radiometabolite into the brain was insignificant (30 min after injection, >96 % of the radioactivity in the target region of the cerebellum was due to the parent compound and only 3–4 % to the radiometabolite). Because 11C-LY2428703 was stable in rat brain homogenates, the presence of radiometabolite in the brain points to its peripheral origin. Moreover, the cerebellar activity ratio of 11C-LY2428703 to that in the forebrain was 3.2–3.5, while the same regional ratio of the radiometabolite was <1.0, suggesting that this radiometabolite had no bioactivity towards the receptor. Therefore, quantitation of binding uptake in the brain can be obtained using a plasma input function.
Dissection followed by LC-MS/MS kinetic analysis of the rat brain with nonlabeled LY2428703 showed that the tracer accumulated preferentially in the target-rich cerebellum relative to the frontal cortex at all time points. This distribution matches the known distribution of mGluR1s in the rat and human brain [19, 20] and indicates high specific binding. Further validation of the biodistribution of the compound would have required autoradiographic studies, which are difficult to perform accurately with 11C labeling. The time-course study showed rapid uptake of the tracer and washout in a time frame suitable for PET ligand 11C imaging.
It is also important to note that, taken together, the evidence from in vitro studies and binding assays showed that the nonlabeled tracer LY2428703 was highly specific and selective. In fact, oral LY2428703 dose-dependently occupied rat mGluR1s, resulting in concentration decreases in the selective mGluR1 antagonist JNJ16259685. JNJ16259685 also blocked the specific binding of LY2428703 when the latter was used as a tracer. Conversely, little to no mGluR5 occupancy by LY2428703 was seen under the same treatment paradigm and using the same animals.
PET studies conducted in rodents showed that 11C-LY2428703 had excellent characteristics for in vivo imaging. It readily penetrated the brain and distributed preferentially in the target-rich cerebellum. The signal was readily decreased by mGluR1 blocking agents, but not influenced by mGluR5 blockers; therefore, further studies in mGluR1 and mGluR5 knockout animals were deemed unnecessary. Moreover, 11C-LY2428703 did not seem to be a substrate for efflux transporters at the blood–brain barrier, as suggested by results of the in vitro MDCK assay and the in vivo PET studies using P-gp and BCRP knockout mice.
Conclusion
11C-LY2428703, a new PET radioligand for mGluR1 quantification, displayed promising characteristics in vitro, ex vivo, and in vivo in rodents.
References
Spooren W, Ballard T, Gasparini F, Amalric M, Mutel V, Schreiber R. Insight into the function of group I and group II metabotropic glutamate (mGlu) receptors: behavioural characterization and implications for the treatment of CNS disorders. Behav Pharmacol. 2003;14:257–77.
Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–37.
Steckler T, Oliveira AF, Van Dyck C, Van Craenendonck H, Mateus AM, Langlois X, et al. Metabotropic glutamate receptor 1 blockade impairs acquisition and retention in a spatial water maze task. Behav Brain Res. 2005;164:52–60.
Ferraguti F, Crepaldi L, Nicoletti F. Metabotropic glutamate 1 receptor: current concepts and perspectives. Pharmacol Rev. 2008;60:536–81.
Ametamey SM, Kessler LJ, Honer M, Wyss MT, Buck A, Hintermann S, et al. Radiosynthesis and preclinical evaluation of 11C-ABP688 as a probe for imaging the metabotropic glutamate receptor subtype 5. J Nucl Med. 2006;47:698–705.
Hamill TG, Krause S, Ryan C, Bonnefous C, Govek S, Seiders TJ, et al. Synthesis, characterization, and first successful monkey imaging studies of metabotropic glutamate receptor subtype 5 (mGluR5) PET radiotracers. Synapse. 2005;56:205–16.
Simeon FG, Brown AK, Zoghbi SS, Patterson VM, Innis RB, Pike VW. Synthesis and simple 18F-labeling of 3-fluoro-5-(2-(2-(fluoromethyl)thiazol-4-yl)ethynyl)benzonitrile as a high affinity radioligand for imaging monkey brain metabotropic glutamate subtype-5 receptors with positron emission tomography. J Med Chem. 2007;50:3256–66.
Huang Y, Narendran R, Bischoff F, Guo N, Zhu Z, Bae SA, et al. A positron emission tomography radioligand for the in vivo labeling of metabotropic glutamate 1 receptor: (3-ethyl-2-[11C]methyl-6-quinolinyl)(cis-4-methoxycyclohexyl)methanone. J Med Chem. 2005;48:5096–9.
Yanamoto K, Konno F, Odawara C, Yamasaki T, Kawamura K, Hatori A, et al. Radiosynthesis and evaluation of [(11)C]YM-202074 as a PET ligand for imaging the metabotropic glutamate receptor type 1. Nucl Med Biol. 2010;37:615–24.
Hostetler ED, Eng W, Joshi AD, Sanabria-Bohorquez S, Kawamoto H, Ito S, et al. Synthesis, characterization, and monkey PET studies of [18F]MK-1312, a PET tracer for quantification of mGluR1 receptor occupancy by MK-5435. Synapse. 2010;65:125–35.
Fujinaga M, Yamasaki T, Kawamura K, Kumata K, Hatori A, Yui J, et al. Synthesis and evaluation of 6-[1-(2-[(18)F]fluoro-3-pyridyl)-5-methyl-1H-1,2,3-triazol-4-yl]quinoline for positron emission tomography imaging of the metabotropic glutamate receptor type 1 in brain. Bioorg Med Chem. 2011;19:102–10.
Yamasaki T, Fujinaga M, Yoshida Y, Kumata K, Yui JJ, Kawamura K, et al. Radiosynthesis and preliminary evaluation of 4-[(18)F]fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide as a new positron emission tomography ligand for metabotropic glutamate receptor subtype 1. Bioorg Med Chem Lett. 2011;21:2998–3001.
Prabhakaran J, Majo VJ, Milak MS, Kassir SA, Palner M, Savenkova L, et al. Synthesis, in vitro and in vivo evaluation of [11C]MMTP: a potential PET ligand for mGluR1 receptors. Bioorg Med Chem Lett. 2010;20:3499–501.
Fujinaga M, Yamasaki T, Yui J, Hatori A, Xie L, Kawamura K, et al. Synthesis and evaluation of novel radioligands for positron emission tomography imaging of metabotropic glutamate receptor subtype 1 (mGluR1) in rodent brain. J Med Chem. 2012;55:2342–52.
Tsui HT, Gillard SE, Argilli E, Zia-Ebrahimi M, Vannieuwenhze MS, Wheeler WJ, et al. (3H)-LY456066, a potent subtype selective radioligand for metabotropic glutamate receptor subtype 1. Society for Neuroscience Meeting. 2002; Abstract no. 247.3.
Raub TJ, Lutzke BS, Andrus PK, Sawada GA, Staton BA. Early preclinical evaluation of brain exposure in support of hit identification and lead optimization. In: Borchardt RT, Middaugh CR, editors. Optimizing the “drug-like” properties of leads in drug discovery. Biotechnology: Pharmaceutical Aspects Series. Berlin: Springer; 2006. p. 355–410.
Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–5.
Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos. 2002;23:327–38.
Mutel V, Ellis GJ, Adam G, Chaboz S, Nilly A, Messer J, et al. Characterization of [3H]quisqualate binding to recombinant rat metabotropic glutamate 1a and 5a receptors and to rat and human brain sections. J Neurochem. 2000;75:2590–601.
Fotuhi M, Sharp AH, Glatt CE, Hwang PM, von Krosigk M, Snyder SH, et al. Differential localization of phosphoinositide-linked metabotropic glutamate receptor (mGluR1) and the inositol 1,4,5-trisphosphate receptor in rat brain. J Neurosci. 1993;13:2001–12.
Acknowledgments
The authors are grateful to the NIH Clinical PET Center (Chief, Dr. P. Herscovitch) for cyclotron production of carbon-11. Ioline Henter provided excellent editorial assistance.
Disclosure
This work was supported in part by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 65.0 KB)
Rights and permissions
About this article
Cite this article
Zanotti-Fregonara, P., Barth, V.N., Liow, JS. et al. Evaluation in vitro and in animals of a new 11C-labeled PET radioligand for metabotropic glutamate receptors 1 in brain. Eur J Nucl Med Mol Imaging 40, 245–253 (2013). https://doi.org/10.1007/s00259-012-2269-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-012-2269-7