
Abstract
Background
[123I]N-ω-fluoropropyl-2β-carbomethoxy-3β-{4-iodophenyl}nortropane ([123I]FP-CIT) single photon emission computed tomography (SPECT) is a frequently and routinely used technique to detect or exclude dopaminergic degeneration by imaging the dopamine transporter (DAT) in parkinsonian and demented patients. This technique is also used in scientific studies in humans, as well as in preclinical studies to assess the availability of DAT binding in the striatum. In routine clinical studies, but also in scientific studies, patients are frequently on medication and sometimes even use drugs of abuse. Moreover, in preclinical studies, animals will be anesthetized. Prescribed drugs, drugs of abuse, and anesthetics may influence the visual interpretation and/or quantification of [123I]FP-CIT SPECT scans.
Discussion
Here, we discuss the basic principle of how drugs and anesthetics might influence the visual interpretation and/or quantification of [123I]FP-CIT SPECT scans. We also review drugs which are likely to have a significant influence on the visual interpretation and/or quantification of [123I]FP-CIT SPECT scans. Additionally, we discuss the evidence as to whether frequently prescribed drugs in parkinsonian and demented patients may have an influence on the visual interpretation and/or quantification of [123I]FP-CIT SPECT scans. Finally, we discuss our recommendations as to which drugs should be ideally withdrawn before performing a [123I]FP-CIT SPECT scan for routine clinical purposes. The decision to withdraw any medication must always be made by the specialist in charge of the patient’s care and taking into account the pros and cons of doing so.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Idiopathic Parkinson’s disease (PD) and several other parkinsonian syndromes are characterized neuropathologically by degeneration of dopaminergic cells, resulting in a loss of dopamine transporters (DATs) in the striatum [1, 2]. Several radiotracers for positron emission tomography (PET) and single photon emission computed tomography (SPECT) have been developed successfully to assess the integrity of presynaptic dopaminergic neurons end postsynaptic receptors in humans, including radiotracers for the DAT (Fig. 1) [3–12]. In the past decade, DAT imaging was shown to be a very sensitive means to detect loss of striatal DATs (particularly in the putamen) in early PD (for reviews, see [13, 14]), and recent studies even suggest that DAT imaging may be able to detect nigrostriatal dopaminergic degeneration in preclinical cases [15, 16]. Furthermore, and in line with autopsy studies, recent studies showed the ability of DAT imaging to differentiate dementia with Lewy bodies (DLB) from Alzheimer’s disease (AD) [17–19]. In DLB, but not in AD, a loss of striatal DAT binding has been found. In clinical practice, it is sometimes difficult, but important, to discriminate parkinsonian patients with dopaminergic cell loss from those with other forms of parkinsonism not characterized by loss of presynaptic dopaminergic cells (e.g., psychogenic parkinsonism or drug-induced postsynaptic parkinsonism) [14]. The same is true for the differentiation between DLB and AD [18, 19]. Following the promising result of the initial SPECT studies in parkinsonian patients using [123I]N-ω-fluoropropyl-2β-carbomethoxy-3β-{4-iodophenyl}nortropane ([123I]FP-CIT) as a marker for the DAT [5, 20], registration studies for [123I]FP-CIT were started in 1996 in Amsterdam. In 1998 and 1999, the results of phase-I and -II studies were published, respectively [21, 22], followed by multicenter phase-III and -IV studies in 2000 and 2004, respectively [23, 24].

Simplified diagram of a striatal dopaminergic synapse. On the presynaptic side, potential markers for imaging of the integrity of dopaminergic neurons are shown. 6-[18F]fluoro-l-3,4-dihydroxyphenylalanine ([ 18 F]DOPA) PET provides a measure of the structural and biochemical integrity of the dopaminergic neurons [3]. The radiotracer is taken up in the dopaminergic neuron via an amine acid transporter and is then decarboxylated to fluorodopamine by l-aromatic acid decarboxylase and temporarily stored in vesicles within the nerve terminals. [11C]dihydroxytetrabenazine ([ 11 C]DTBZ) is a commonly used marker for the vesicular transporter [vesicular monoaminergic transporter-2 (VMAT-2)] in humans [4]. Substituted (nor)phenyltropanes ([ 123 I]FP-CIT, [ 123 I]β-CIT, [ 11 C]PE2I, [ 11 C]CFT, and [ 99m Tc]TRODAT) are frequently used PET and SPECT tracers for imaging of DAT in humans [5–9]. Dopamine D2 receptors are much more expressed on the postsynaptic side than on the presynaptic side of the dopaminergic synapse. Commonly used radiotracers for D2/3 receptors are substituted benzamides ([ 123 I]IBZM, [ 11 C]raclopride, and [ 18 F]fallypride) [10–12]. For convenience, only D2 receptors are shown on the postsynaptic cell, whereas other dopaminergic receptors (e.g., D1 receptors) are also located on this side. DAT dopamine transporter, VMAT-2 vesicular monoaminergic transporter-2
In 2000, 123I-FP-CIT was licensed as DaTSCAN™ in Europe to differentiate patients suffering from parkinsonian syndromes, such as PD, multiple system atrophy, or progressive supranuclear palsy, from essential tremor (ET). In 2006, the same product received an additional registration for a new indication: to differentiate scintigraphically DLB from AD.
After registration in 2000, the use of [123I]FP-CIT has increased dramatically. In 2006, more than 500 centers in 30 countries throughout Europe have been using [123I]FP-CIT, both for routine clinical and experimental studies in animals and humans [25–29]. Nowadays, [123I]FP-CIT is used extensively in clinical practice for the investigation of parkinsonian patients, as well as in experimental studies, and it is reasonable to expect that the use of this radiotracer will increase even further in the near future to discriminate DLB from AD. However, it is of paramount importance to evaluate potential effects of drugs on DAT imaging with [123I]FP-CIT. Therefore, in this review, we screened the literature to evaluate which kinds of drugs (of abuse) are likely to influence [123I]FP-CIT SPECT imaging.
In the first part of this review, we constructed a theoretical framework to improve the understanding of potential influences of drugs on [123I]FP-CIT SPECT images. In the second part, we described extensively what kind of medication will influence [123I]FP-CIT SPECT images, based on studies performed with [123I]FP-CIT or other radiotracers for the DAT. In addition, the possible influence of all groups of drugs on [123I]FP-CIT SPECT imaging that are prescribed frequently in parkinsonian or demented patients will be discussed. In the third part, we describe the way we propose to deal with the information put forward in parts 1 and 2, both in an experimental and a routine clinical setting, although focused on the last setting.
Part 1: how could drugs theoretically influence the results of [123I]FP-CIT SPECT images
Drugs with affinity for monoaminergic transporters, or that may influence the affinity of [123I]FP-CIT for monoaminergic transporters
[123I]FP-CIT is a 123I-labeled tracer for the DAT and is derived from cocaine [30]. Its in vitro affinity for the DAT is high (approximately 2 nM) ([31] please note that, in this particular paper, [123I]FP-CIT is named RTI-313). The affinity for the serotonin transporter (SERT) is moderate, while the affinity for the norepinephrine transporter (NET) is low (16 and 140 nM, respectively) [31]. Based on these data, it is likely that the in-vivo binding of [123I]FP-CIT is primarily to DAT and to a lesser amount to SERT. Consequently, drugs with high affinity for the DAT and SERT (such as reuptake inhibitors), but presumably not for the NET, may influence [123I]FP-CIT images directly.
Within the striatum, the concentration of DAT is much higher than for SERT [32]. In line with this, initial biodistribution studies in rats [33] showed that striatal [123I]FP-CIT uptake could be blocked with a selective ligand for the DAT (GBR12909), but not by a selective SERT blocker (fluvoxamine). Indeed, also human DAT imaging studies showed that drugs with (relatively) high affinity for the DAT influence radiotracer binding in the striatum. For example, the amphetamine-like drug methylphenidate blocks the specific striatal binding of [123I]FP-CIT significantly [34].
Theoretically, a drug that affects the apparent affinity of [123I]FP-CIT for the DAT or SERT might induce changes in striatal [123I]FP-CIT binding ratios. For example, radiotracers for the dopamine D2 receptors could be displaced by a large amount of endogenous dopamine (e.g., induced by amphetamines), which, among others, is reflected in an increase of the radioligand K d for the D2 receptor [35]. It is remarkable that acute administration of amphetamine in monkeys induced a 50% decrease in striatal 2β-carboxymethoxy-3β-(4-iodophenyl)tropane (β-CIT) binding [32], but in that study, the reason for this decrease was not studied. However, other studies showed that amphetamines induced fast internalization of DATs, which will induce a decrease in the number of transporters (Bmax) but not in affinity (K d) [36]. On the other hand, it has been suggested that high plasma levels of the adrenergic agonists phenylephrine or norepinephrine may change the apparent affinity of radiotracers for the DAT. This suggests that drugs which increase plasma levels of phenylephrine or norepinephrine might be able to affect striatal binding of [123I]FP-CIT [37].
In human studies, striatal [123I]FP-CIT binding is, in the vast majority of studies, quantified as (specific) striatal binding over nonspecific binding in the occipital cortex or cerebellum. One has to take into account that, although the concentration of SERT is low in the striatum, occipital cortex, and cerebellum, it is not negligible [38]. Therefore, blockade of FP-CIT binding to SERTs by drugs may lead, at least theoretically, to an increase in the binding ratio because changes in the denominator of the ratio can induce larger changes in the value of the ratio than changes in the numerator. Interestingly, studies in rats showed that [123I]FP-CIT binding in the occipital cortex could be displaced by a selective serotonin reuptake inhibitor (SSRI), but not by a selective blocker for the DAT [33]. Moreover, ecstasy (XTC) is a selective neurotoxin for serotonergic cells and consequently induces loss of SERT. Interestingly, DAT studies in XTC users showed significantly higher striatal-to-cerebellar [123I]β-CIT binding ratios as compared to controls [39]. Taking all these data together, it may be suggested that DAT and SERT blockers (e.g., cocaine or SSRIs) and substrates for these transporters (e.g., amphetamines) may influence striatal [123I]FP-CIT binding ratios.
Drugs that may influence the expression of DAT and SERT
As discussed in the previous section, [123I]FP-CIT primarily labels in vivo the DAT, but also to some extent the SERT. The expression of DAT and SERT is extremely complex and dynamic [40, 41]. The DAT and SERT play critical roles in terminating dopaminergic and serotonergic transmission by reuptake of dopamine and serotonin from the synaptic cleft, respectively. Control of DAT and SERT activity and expression are therefore central to the spatial and temporal regulation of synaptic dopamine and serotonin levels. DATs and SERTs rapidly traffic between the plasma membrane and endosomal compartments in both constitutive and protein kinase C (PKC)-dependent manners [42]. Kinase activators, phosphatase inhibitors, and substrates for the transporter modulate DAT and SERT phosphorylation (and consequently modulate expression on the cell membrane) and activity, but the underlying mechanisms and role of phosphorylation in these processes are poorly understood [43–46]. However, it is well accepted that phosphorylation of DAT or SERT, by activation of PKC or inhibition of protein phosphatases, will lead to reduction of transporter expression on the cell membrane. Therefore, at least theoretically, drugs which influence PKC (e.g, via activation of presynaptic receptors located on DAergic neurons) [47], influence phosphatase inhibitors, or are substrates for the transporter may induce alterations in the expression of the DAT and/or SERT and, consequently, may influence quantification of these transporters.
Potential effects of drugs on the metabolism of [123I]FP-CIT and on cerebral blood flow
The main metabolic pathway of cocaine is hydrolysis of the two ester functions, resulting in the formation of benzoylecgonine and ecgonine methyl ester [48, 49]. Hydrolysis of the 2β-caroboxymethyl-ester function, spontaneously and by esterases, is also the main metabolic pathway for [123I]FP-CIT (Fig. 2), leading to [123I]FP-CIT-acid [50]. However, FP-CIT-acid is probably not lipophilic enough to pass the blood–brain barrier [51], and it is therefore unlikely that drugs that do influence esterase activity will be able to influence [123I]FP-CIT binding ratios to central DATs or SERTs. A second, minor metabolic pathway in cocaine bioactivation includes N-demethylation by microsomal cytochrome P-450. Also, for [123I]FP-CIT, N-demethylation by microsomal cytochrome P-450 occurs in vivo in humans [51] (Fig. 2). Indeed, a radiotracer is then formed, called nor-β-CIT. Previous studies showed that the affinity for SERT is higher for nor-β-CIT than for [123I]FP-CIT [52]. It is known that, in healthy volunteers, less than <4% of the total amount of activity is nor-β-CIT 1 h after injection of [123I]FP-CIT [51]. Therefore, medication that influences cytochrome P-450 activity might influence the metabolism of [123I]FP-CIT. However, one has to take into account that many isoenzymes of P-450 (CYP) exist. Because only CYP3A plays a significant role in the N-demethylation of cocaine [53] and consequently presumably also in the N-demethylation of [123I]FP-CIT, only medications that influence CYP3A are potential influencers of the metabolism of [123I]FP-CIT.

Metabolism of [123I]FP-CIT in humans. Metabolism I represents the hydrolysis of ester function which leads to [123I]FP-CIT-acid, the major metabolite found in plasma (>60% at 2 h p.i. [50]). Metabolism II represents the N-dealkylation of the radiotracer by P-450 cytochrome, which leads to the formation of [123I]nor-β-CIT, a minor metabolite (<4% [51])
Finally, in human [123I]FP-CIT SPECT studies, acquisition is started 3–6 h after injection of the radiotracer [22] because, in that time period, the specific striatal binding ratio is stable and a so-called pseudoequilibrium is reached. The relative slow kinetics of FP-CIT approximates the kinetics characteristics of a radiotracer with irreversible uptake [54]. It is known for radiotracers in which binding is irreversible that a change in binding is positively correlated with a change in cerebral blood flow [55]. Therefore, at least theoretically, a drug which influences cerebral blood flow may affect striatal FP-CIT binding ratios.
Part 2: groups of drugs and their potential influence on [123I]FP-CIT SPECT imaging
Definition of influence
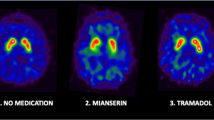
[123I]FP-CIT is used in many centers in Europe for routine clinical studies. In most of these centers, FP-CIT SPECT scans are examined visually, based on the results of reports that show that visual examination of [123I]FP-CIT images is a sensitive means to differentiate parkinsonian diseases with, from those without, dopaminergic degeneration [23]. In a typical early PD case, there is asymmetrical striatal [123I]FP-CIT uptake, with severe loss of binding in the putamen, especially at the contralateral side, resulting in a “full stop” sign (at least at one side), while in ET, the “comma” sign, representing symmetrical binding, both in caudate and putamen, is present (Fig. 3). Theoretically, visual assessment of [123I]FP-CIT could be influenced by drugs in at least five ways: (1) A drug blocks [123I]FP-CIT uptake in striatum (both in caudate and putamen) or lowers the numbers of DATs or the affinity of [123I]FP-CIT for the DAT, resulting in an underestimating of the real number of striatal DATs, which may result in a misleading scan. (2) A drug reduces only uptake of [123I]FP-CIT in the putamen or caudate nucleus, resulting in an abnormal scan. There is some indication that transporters may be differentially regulated in certain brain regions. Specifically, DAT regulation may differ in the dorsal striatum from that in the nucleus accumbens (part of the ventral striatum) [47]. Although it may be hard to differentiate FP-CIT binding in the caudate nucleus from that in the nucleus accumbens, it is important to note the volume of accumbens is much smaller than that of the caudate [56] and that the dorsal striatum contributes to 70–80% of the measured SPECT signal, as opposed to only 20–30% contribution of the ventral striatum [57]. Therefore, this is an unrealistic mechanism to induce a misleading abnormal FP-CIT scan. (3) A drug induces increases striatal [123I]FP-CIT uptake (both in caudate and putamen), or lowers background activity, which induces a better contrast of striatal uptake vs background activity. However, in such a situation, a normal scan will still be judged as normal, and a “full stop” sign in, e.g., a PD patient will not change to a “comma” sign. (4) A drug induces an increase of [123I]FP-CIT uptake in the putamen, but not in the caudate nucleus, or vice versa. However, this is only a theoretical possibility, not supported by data from literature. (5) A drug induces a significant change in rCBF.

Transversal [123I]FP-CIT SPECT images at the level of the striatum, in early PD (left panel) and in ET (right panel). Left panel: note the asymmetric binding, with less binding in the right vs the left striatum. At the right side, binding is only visible in the caudate nucleus (full stop sign), while at the left side, binding is visible in the caudate nucleus and anterior putamen. Right panel: intense binding both in the caudate nucleus and putamen, bilaterally (comma sign)
From this list of five theoretical ways that a [123I]FP-CIT SPECT image could be influenced visually, the first one may be the most relevant one. For example, if a drug blocks 90% of specific striatal [123I]FP-CIT uptake, it will be hard to visualize striatal uptake at all and it may lead to a misleading scan. However, because it has not been evaluated how many DATs should be blocked or down-regulated before a “normal” [123I]FP-CIT scan might be misdiagnosed as an “abnormal” scan, it may be reasonable to assume that a more than 20% blockage of striatal DATs by drugs may induce a misleading scan (in case of visual assessment). Although 20% is an arbitrary threshold, it is a conservative one, giving the large variation in striatal DATs in a healthy control population [58].
In several institutions, [123I]FP-CIT SPECT images are not only analyzed visually but also quantitatively, most frequently with a region-of-interest technique. From test/retest studies in healthy controls, it is known that the reproducibility is, on average, approximately 7% (SD 3%) when the scans were analyzed with a template with fixed regions for the striatum [59]. Therefore, it is reasonable to assume that for an individual patient in a routine clinical setting, a drug should have an effect on [123I]FP-CIT binding ratios in the striatum that is larger than the test–retest measurements plus twice the SD or, in other words, larger than 13%. For scientific studies, all potential effects of drugs should be taken into account, as well as effects that may be within the reproducibility of the test, especially when there are small differences in striatal DAT densities between the groups under study, or if serial studies are performed in which a drug has been introduced, or as the dose has been changed after the baseline scan.
In this review, we will discuss the potential effects of drugs on the visual assessment and quantification of [123I]FP-CIT SPECT scans. We will discuss drugs that are likely to influence FP-CIT SPECT scans, and we will also discuss the group of drugs that are frequently used in parkinsonian and demented patients in relation to their likelihoods to influence FP-CIT SPECT scans.
Effects of CNS stimulants, including DAT blockers and sympathicomimetics
As [123I]FP-CIT is derived from cocaine, it is straightforward that acute administration of cocaine will substantially occupy DATs in the striatum and consequently shall influence striatal [123I]FP-CIT uptake. Although chronic cocaine intake may initially induce an upregulation of DATs (via a rapid recycling of internalized DATs to the membrane surface) [60, 61], the blocking effects after administration will overrule this effect and will have a major effect on DAT imaging. Volkow and coworkers [62] already showed in their seminal paper in 1997 that acute administration of cocaine in doses used by addicts induces a high percentage of blockage of striatal DATs. In fact, only if 50% of DATs are blocked, a “high” is experienced by the cocaine abuser [62]. Therefore, acute administration of cocaine will not only influence quantification of DATs but will also influence the visual examination of [123I]FP-CIT SPECT images (Table 1).
Amphetamines like methamphetamine or dexamphetamine are not only drugs of abuse. For example, d-amphetamine is a frequently prescribed drug as an appetite suppressant or in attention-deficit-hyperactivity-disorder (ADHD) patients. These amphetamines have a relatively low affinity for DATs [Ki in the micromolar range; 52], but maybe more importantly, these drugs are substrates for the DAT and may induce fast internalization of DATs (and thereby reduce the surface expression to bind the FP-CIT) presumably via the PKC system [36]. For example, Laruelle and coworkers [32] have shown in monkeys that approximately 50% of striatal β-CIT binding is displaced by amphetamine, maybe by a fast internalization. Moreover, recent DAT SPECT and PET studies have shown that patients on methamphetamine or with a short abstinence period for methamphetamine (less than 6 months) had significantly lower striatal binding ratios than controls, ranging from 20 to 30% [63, 64]. Furthermore, several reports have shown that prolonged use of these drugs may have long-lasting effects on the expression of the DAT [65–67], although this phenomenon has been debated by the results of studies performed in methamphetamine abusers with protracted abstinence [66]. Nevertheless, acute or recent administration of these drugs will influence [123I]FP-CIT SPECT quantification and visual assessments of images (Table 1) and may be an example of how a drug could influence the result of a [123I]FP-CIT SPECT image by influencing the expression of the DAT.
Methylphenidate is also a drug derived from amphetamine and frequently prescribed for ADHD and narcolepsy. Vles et al. [34] reported recently that a therapeutic dose of methylphenidate decreased striatal [123I]FP-CIT binding ratios up to 75% both in the putamen and the caudate nucleus. Moreover, a recent study showed that not only the immediate release form of methylphenidate but also the osmotic release form may induce a large occupancy of striatal DATs [68]. All in all, recent administration of methylphenidate is likely to influence the visual assessment of [123I]FP-CIT SPECT scans, as well as the quantification of DATs by FP-CIT SPECT (Table 1), and it may be a typical example of how a drug with affinity for the DAT could influence the result of a [123I]FP-CIT SPECT image.
Like amphetamines, the CNS stimulants (nor)ephedrine, pseudoephedrine, and phentermine are structurally similar to methamphetamine and were or are commonly used appetite suppressants. Chronic ephedrine use, in nutritional supplements, has been reported in female weightlifters [69], and ephedrines are frequently used as an ingredient in widely marketed herbal preparations. Phentermine is sometimes prescribed to induce weight loss. As compared to amphetamines, typical clinical doses of phentermine and ephedrines may not release central DA in humans [70]. Although the most potent actions of ephedrine-type compounds were as substrates of the NET transporter, some ephedrine derivatives showed affinity for the DAT in the low nanomolar or micromolar range, or they are substrates for the DAT [71–73]. Ephedrine and pseudoephedrine are also over-the-counter sympathomimetics to be used as bronchodilatators and nasal decongestants, respectively. Unfortunately, the influence of these drugs on DAT imaging has not been studied. However, taking all data together, we could not exclude an effect of these drugs on DAT studies, particularly when used as tablets. However, it is unlikely that ephedrine-like drugs used as bronchodilatators or nasal decongestants will significantly influence central DAT imaging because the plasma concentrations will be too low.
The combination of the amphetamine-derivatives fenfluramine (5-HT releaser) plus the DA releaser phentermine (fen-phen) has also been used widespread as anorectics to treat obesity. The adverse effects that came to be associated with fenfluramine and dexfenfluramine, valvular heart disease and primary pulmonary hypertension [74, 75], have led to their eventual withdrawal from the market in 1997 [76]. However, nowadays, fenfluramine is still used illicitly. Initial studies in small laboratory animals showed that acute administration of fenfluramines induced an increase in striatal DAT [77], which is remarkable because it is also known to increase PKC activity [78]. Fenfluramines (i.e., racemic fenfluramine and dexfenfluramine) cause dose-related, long-lasting reductions in serotonin axonal markers in all the animal species tested and with all the routes of drug administration used. Doses of fenfluramines that produce signs of brain serotonin neurotoxicity in animals are on the same order as those used to treat humans for weight loss when one takes into account known relations between body mass and drug clearance [79]. This may lead to lower FP-CIT binding to SERTs in fenfluramine users and, consequently, may influence FP-CIT DAT binding ratios. However, no human DAT imaging data on this topic are available, and therefore, it is still unknown if fenfluramine influences DAT imaging in humans.
Modafinil is a novel wakefulness-promoting agent that has been shown to have greater efficacy than placebo in the treatment of ADHD and narcolepsy. Modafinil has been shown to have a low affinity for the DAT [80]. However, the finding that modafinil and amphetamine induce similar increases in dopamine release at equipotent wake-promoting doses suggest a role for the DAT [81]. Indeed, a recent study showed that modafinil is able to block the DAT at “therapeutic doses” substantially in monkey brain [82]. Therefore, just like the other abovementioned drugs, this amphetamine analog may induce significant changes in [123I]FP-CIT quantification and, possibly, also in the visual interpretation (Table 1).
The NET and DAT blocker mazindol is sometimes prescribed for the treatment of obesity or depression. A recent [123I]β-CIT SPECT study in human cocaine abusers suggests that low doses of mazindol (i.e., 2–4 mg) occupy approximately 25% [83]. Therefore, this drug may affect [123I]FP-CIT imaging, both visually and quantitatively (Table 1).
Bupropion is frequently prescribed as an antidepressant or as an antismoking drug. Several reports have shown that bupropion blocks DATs in vivo, although the results are not consistent. For example, a recent DAT SPECT study showed that after 4 weeks of bupropion in nine depressed patients, a 20% decrease in striatal DAT binding ratios was induced [84]. In addition, another PET study in depressed patients reported that the occupancy after bupropion treatment was 14% [85]. Although this effect may not necessarily indicate an effect of bupropion on DAT per se, a recent PET study performed in healthy controls showed that, 3 h after the last dose of bupropion SR, average DAT occupancy by bupropion and its metabolites was 26%. This level of occupancy was maintained through the last PET assessment at 24 h after dosing [86]. In contrast to this, another study performed in healthy controls found no effects of bupropion on DAT binding [87]. Nevertheless, although the effects of bupropion on DAT imaging are not consistent, a possible effect could not be excluded, and this effect may be approximately 20% and, thus, may influence visual and quantitative analyses of [123I]FP-CIT SPECT studies (Table 1).
Byas-Smith and coworkers [37] showed in anesthetized monkeys that striatal DAT binding as assessed with 8-(2-[18F]fluoroethyl)-2β-carbomethoxy-3β-(4-chlorophenyl)nortropane (FECNT) PET increased by approximately 50% during the phenylephrine or norepinephrine infusion. By performing additional in vitro studies, the authors were also convinced that a change in affinity of [18F]FECNT for the DAT (rather than a change in DAT density) occurs during these manipulations. These data not only show that the DAT is considerably dynamic in its regulatory capacity but that sympathomimetics do have a potential effect on DAT imaging possibly by influencing the apparent affinity of the radiotracer for the DAT. However, one has to take into account that, in these experiments, the infusion rates were increased until the blood pressure increase precipitated a significant bradycardia or the mean arterial blood pressure increased by 25% [37]. Therefore, future studies are needed to study whether or not drugs used at clinical doses, which increase levels of phenylephrine or norepinephrine, do affect DAT imaging.
Effects of antidepressants
Recent studies using [123I]β-CIT SPECT to label DATs in vivo showed that the use of SSRIs, as well as the use of a selective serotonergic neurotoxic (XTC), induced a significant increase in the striatal [123I]β-CIT uptake ratios [87–89]. However, β-CIT (or RTI-55) has a higher affinity and less selectivity for the SERT than [123I]FP-CIT [31]. Interestingly, we recently showed that, while one dose of 20 mg of citalopram leads to a significant increase of β-CIT striatal binding ratios of approximately 20%, two doses of 20 mg paroxetine leads to less increase of striatal [123I]FP-CIT SPECT (approximately 10% [90]). This finding is in line with the observation that [123I]FP-CIT is a more selective radiotracer for the DAT than β-CIT. In this study, paroxetine has been used as a SERT blocker. Although we have no direct evidence that other SSRIs will have the same effects on striatal FP-CIT binding ratios, it is likely that other SSRIs will also show the same effect. Yet, within the group of SSRIs, one of the SSRIs has a relatively high affinity for the DAT that is sertraline (affinity for DAT approximately 20–25 nM) [91, 92]. It could be hypothesized that the DAT blocking effects of sertraline on striatal FP-CIT binding ratio are counterbalanced by its SERT blocking effects. However, initial studies in mice did not confirm this hypothesis by showing a significant increase of striatal β-CIT binding after administration of sertraline [93]. All in all, although there is evidence to suggest that at least one SSRI significantly influences the quantification of FP-CIT to DAT in humans, these effects are too small to hinder the interpretation of visual assessments and will lead to increases of striatal to occipital ratios of approximately 10%.
Serotonin–norepinephrine reuptake inhibitors (SNRIs) (e.g., venlafaxine and clomipramine) are in use today for the treatment of anxiety disorders and depression. Studies in mice and rats [94, 95] showed that clomipramine induced an increase in striatal β-CIT binding comparable to the situation with SSRIs. Moreover, Shang and coworkers recently showed a significantly 10% increase of striatal β-CIT ratio by venlafaxine in humans [96]. Therefore, it is likely that the same statements on their potential effects on [123I]FP-CIT imaging could be made for SNRIs as for SSRIs (see above).
Reversible monoamine oxidase-A inhibitors like befloxatone or moclobemide are frequently prescribed for depression. These drugs increase serotonin levels by inhibition of its breakdown, but they do not have a high affinity for the DAT or SERT. Although the effects of these kinds of drugs on DAT imaging have not been evaluated, it is not very likely that they will influence FP-CIT binding to SERTs and indirectly to DATs.
Antidepressants like mirtazapine, buspirone, and nefazodone are α2-adrenergic antagonists, 5-HT1A receptor agonist, and antagonists of 5-HT2C receptors, respectively. While mirtazapine did not increase PKC activity, nefazodone did [78]. However, until now, no studies have evaluated the effects of these drugs on DAT imaging.
Bupropion or mazindol are prescribed in several countries as antidepressants. As described earlier, these drugs may influence DAT imaging. Moreover, radafaxine is a new antidepressant that blocks DAT (and NET) in vivo significantly. In healthy controls, peak blockade of striatal DAT (as assessed with [11C]cocaine PET) occurred at about 4 h after oral intake and was 22% [97]. Based on these data, it is likely that this drug will also decrease striatal FP-CIT binding ratios significantly (Table 1). On the other hand, “older” tricyclic antidepressants such as amitryptiline, imipramine, or mianserine are not potent at the DAT [92], and they are not PKC activators [78]. One study, however, suggested that subchronic treatment (for 10 consecutive days) with desipramine in rats induced a significant increase in striatal DATs [98]. However, unfortunately, DATs were measured with the nonselective DAT tracer [3H]mazindol; therefore, it is not clear if the effects are specific for the DAT. Likewise, Thibaut and coworkers [99] found increased striatal [3H]mazindol uptake in mice after administration of desipramine. They suggested that this phenomenon occurred due to the enhanced availability of radioligand as a consequence of displacement from cerebellar NET binding by desipramine. Overall, although the effects of tricyclic antidepressants on DAT imaging in humans have not been evaluated, it is unlikely that these kinds of drugs will influence DAT imaging significantly.
Effects of neuroleptics
Of the group of neuroleptics, only pimozide (K d = 69 nM) and ziprasidone (K d = 76 nM) had notable potency at the human DAT [100]. In addition, only clozapine induced changes in the PKC level; more specifically, it decreased its level [101]. Until now, one human DAT imaging study specifically examined the effects of a neuroleptic on [123I]FP-CIT binding to DATs. Mateos and coworkers [102] showed in schizophrenic patients that 4 weeks of treatment with risperidone did not influence striatal FP-CIT binding ratios significantly. Moreover, [123I]FP-CIT SPECT studies in schizophrenic patients showed no difference in striatal uptake between drug-free patients and patients on neuroleptics [29], and acute as well as subacute administration of different neuroleptics did not influence striatal FP-CIT binding in rats [103]. These data suggest that if neuroleptics will induce changes in DAT imaging, such changes will presumably not be large enough to influence quantification or visual assessments of [123I]FP-CIT SPECT in routine clinical studies.
Effects of cholinergic and anticholinergic drugs
Anticholinergic drugs are frequently used in parkinsonian patients, and cholinesterase inhibitors (functional cholinergic agonists) are frequently used in demented parkinsonian patients, including DLB patients [104, 105]. Tsukada and coworkers [106] showed that cholinergic neuronal modulations affect striatal DAT activity in the conscious monkey brain. In their study, they showed lower binding potential after acute intravenous administration of the cholinesterase inhibitor donepezil. Importantly, a recent FP-CIT SPECT study compared striatal FP-CIT uptake ratios between large groups of patients on cholinesterase inhibitors with those not on cholinesterase inhibitors and found no significant differences. The results of this study suggest that the use of cholinesterase inhibitors may not influence striatal FP-CIT binding ratios significantly in humans [107]. Therefore, although a small influence of cholinesterase inhibitors on DAT imaging in humans could not be excluded, it is unlikely that cholinesterase inhibitors will significantly influence the interpretation of [123I]FP-CIT SPECT scans in a routine clinical setting.
Kilbourn and coworkers [108] examined DAT binding in rat brain with and without prior intravenous administration of the anticholinergic drug scopolamine (muscarinic receptor blocker; 5 mg/kg body weight). Drug-treated animals exhibited a 30% increase in d-threo-[3H]methylphenidate binding to the DAT in the striatum relative to controls. Also, Tsukada et al. showed a significant increase of DAT binding in monkeys after intravenous scopolamine administration. In parkinsonian patients, anticholinergic drugs, particularly orphenadrine, benztropine, or trihexyphenidyl, are frequently used in particularly younger patients because their psychotoxic, cognitive, and autonomic adverse events make them inappropriate for the treatment of the elderly [109]. Particularly benztropine has a modest affinity for the DAT [110], while the affinity of trihexyphenidyl is in the low micromolar range. Interestingly, a recent study showed that the benztropine analog difluoropine (this is a drug not on the market for human use) occupied DATs up to 76% in monkeys [110]. Consequently, it may be that benztropine induces a lower [123I]FP-CIT binding to DAT due to its potency to occupy the DAT, which influences the quantification and visual examination of FP-CIT SPECT studies. On the other hand, other anticholinergics such as scopolamine (sometimes used as plaster for the prevention of motion sickness), orphenadrine, or trihexyphenidyl may induce an increased [123I]FP-CIT binding to striatal DATs, which may influence quantification but not the visual assessment of scans (see part 2 “Definition of influence”).
Effects of levo-dopa, catechol-O-methyltransferase-inhibitors, monoamine oxidase type B inhibitors, dopamine agonists, and NMDA antagonists
Although the availability of drugs for treatment of PD has multiplied, l-dopa, in its fourth decade of clinical use, is still the most potent and effective medication [111]. Several studies have examined the acute and subchronic effects of levo-dopa on DAT imaging, and the majority of them did not find a significant effect (for a review see [112]). Moreover, the results of a recent study suggested that chronic treatment in PD did not influence [123I]FP-CIT binding. Schillaci and coworkers [113] studied 15 PD patients under stable levo-dopa/carbidopa monotherapy and after at least 20 days of treatment wash-out, and they did not find a significant difference. Therefore, it is unlikely that levo-dopa will have a significant influence on DAT imaging, visually as well as quantitatively. On the other hand, the Parkinson Study Group [114] studied the effects of levo-dopa on the progression of PD. At baseline, PD patients were scanned with [123I]β-CIT SPECT to measure striatal DATs and subsequently treated with levo-dopa at different doses or placebo for 40 weeks. After 40 weeks of treatment, the patients were rescanned (while on treatment) and clinically examined after drug withdrawal for 2 weeks. While the severity of PD increased more in the placebo than in the levo-dopa-treated groups, the decline in striatal DAT binding was significantly greater with levo-dopa than with placebo [−4 to −7.2% among those receiving levo-dopa (dose-dependent effect), as compared with −1.4% among those receiving placebo]. As stated by the authors, they could not exclude the possibility that levo-dopa simply down-regulates the DAT, which may illustrate that drugs may not interfere significantly with [123I]FP-CIT SPECT imaging in routine clinical cases; effects on scientific studies could not be excluded.
The short plasma half-life limits the antiparkinsonian efficacy of levo-dopa/carbidopa. Coadministration of a catechol-O-methyltransferase (COMT) inhibitor extends the plasma half-life of levo-dopa. Therefore, COMT inhibitors are sometimes used in parkinsonian patients, particularly in the more advanced disease stages. Effects of these groups of drugs on DAT imaging have not been studied, but because levo-dopa per se does not significantly interfere with the interpretation of FP-CIT SPECT scans in routine clinical studies, it is unlikely that COMT inhibitors will do so.
The effects of monoamine oxidase type B inhibitors on DAT binding in PD patients have been examined by two groups. Innis and coworkers [115] and Fowler and coworkers [116] showed no significant effects of selegeline on striatal DAT binding in PD patients and healthy controls, respectively. This is remarkable because selegiline is metabolized into amphetamine and methamphetamine [117].
Dopamine D2 agonists, such as pramipexol or pergolide are also frequently prescribed in PD [109, 111]. Several DAT studies examined the subchronic effects of DA agonists in PD patients. While three studies did not find a significant effect [114, 118, 119], one study showed a slight but significant (7%) decrease in striatal DAT binding [120]. These data indicate that DA agonists will not influence the examination of [123I]FP-CIT images visually or quantitatively in routine clinical studies.
The NMDA antagonist amantadine is frequently used in PD, while memantine is used in patients suffering from dementia [121]. Human DAT imaging has not been performed to assess the effects of these drugs on DAT binding. However, binding assays with [3H]GBR-12935 on membranes prepared from animals treated with amantadine revealed no difference in the density and the affinity of striatal DAT binding sites as compared to control, which may indicate that there was no modification at the level of the DAT expression [122]. In contrast, Gordon and coworkers [123] showed an increase of 25–30% of DATs in rats after 3 weeks of intraperitoneal treatment with amantadine.
Effects of estrogen replacement therapy
A growing body of research has shown that the brain dopaminergic system is modulated by estrogen and other sex steroids (for a review, see [124]). Recently, Best and coworkers [125] studied the influence of the menstrual cycle on the DAT availability in humans with β-CIT SPECT. Ten female subjects aged 18–40 years were scanned twice during the early follicular and the midluteal phases to detect any hormone-mediated changes in DAT availability in the striatum. In the 10 menstrual cycle subjects, DAT availability in the striatum did not differ between follicular and luteal phases. Interestingly, Gardiner and coworkers [126] studied 13 postmenopausal women who were administered estrogen replacement therapy and underwent DAT imaging with [99mTc]TRODAT-1 SPECT. In this 6-week pilot study, subjects underwent SPECT before estrogen replacement therapy, after 4 weeks of 0.625 mg/day of conjugated estrogens, and after an additional 2 weeks of 0.625 mg/day CEE plus 10 mg/day of medroxyprogesterone acetate. They showed that short-term administration of estrogen replacement therapy in postmenopausal women is associated with a modest increase in DAT in the putamen but not in the caudate nucleus. Because these effects are small, it is not likely that estrogen replacement therapy will significantly influence the visual interpretation of FP-CIT SPECT scans nor the quantification in routine clinical studies. Finally, the authors’ findings are in contrast to our earlier remarks that it is unlikely that within the dorsal striatum the regulation of the DAT is region-specific (see earlier remark in the paragraph on the “Definition of influence,” first paragraph part 2).
Effects of analgesics (opioids)
Opioids can activate at least four types of opioid receptors (δ, κ, μ, and σ). Activation of opioid receptors may induce changes in striatal DAT densities in rats [127]. It has been shown that the opiate fentanyl (selective μ receptor agonist, frequently used transdermally as an analgestic) may cause reduced [123I]β-CIT binding to striatal DAT [128] in humans (case report; after intrathecal administration) and after acute intraperitoneal administration in rats, although the mechanism is not fully understood. Furthermore, the opioid analgesic meperidine (sometimes used as injection to reduce pain) has atypical opioid receptor agonist effects and shares some structural features with the phenyltropane analogs of cocaine [129]. In addition, meperidine appears to interact predominantly with the high-affinity component of the DAT, although with relatively low affinity (but relatively high affinity for the SERT [130]). On the other hand, the IC50 to inhibit DA uptake is relatively high [131]. Interestingly, Xiao and coworkers [132] showed in a DAT SPECT study in rhesus monkey that acute morphine (agonist for the μ and κ receptor) injection has both rapid and lasting effects on DAT by down-regulating its function. The decline was partially reversible following morphine abstinence. On the other hand, chronic but not acute treatment of rats with morphine significantly decreased DAT density in the anterior basal forebrain that includes the nucleus accumbens but had no such effect on binding in the striatum [133]. Additionally, Kish and coworkers studied biochemical indices of monoaminergic neurotransmitter activity and integrity in postmortem striatum of chronic heroin users who died from an overdose of heroin. Striatal levels of the vesicular monoamine transporter were normal, suggesting that the density of dopamine nerve terminals is not reduced in heroin users [134]. All in all, it cannot be excluded that opiates influence binding of [123I]FP-CIT imaging to DATs, but data are not consistent.
Effects of anesthetics
PET and SPECT studies in animals play an important role in the characterization of new radiotracers, and these studies present an in vivo means to measure neuroreceptors in animal models of disease or to evaluate treatment. Particularly, in the past few years, PET and SPECT imaging of small laboratory animals has been improved substantially (for reviews see [135, 136]). In the vast majority of such studies, the animal will be anesthetized. Particularly, the famous PET studies in conscious monkeys by Tsukada’s group have highlighted the potential effects of anesthetics on DAT imaging. For example, these studies showed clear effects of ketamine and isoflurane on DAT imaging [137, 138], presumably by a fast internalization of DATs from the cell membrane and/or blockade of DAT binding [139, 140]. These data are relevant for the interpretation of animal studies in which [123I]FP-CIT was used to label the DAT. For clinical studies, this information may be of relevance because ketamine is sometimes used as a drug of abuse [141] (Table 1). Similar to ketamine, the dissociative anesthetic phencyclidine (PCP) may exert some direct effects through the DAT [142], and it is sometimes used as a drug of abuse (Table 1).
Part 3: drugs interactions in imaging DAT with FP-CIT in routine clinical studies: recommendations for withdrawal of drugs before the scan
In scientific studies, even small differences may be relevant. Therefore, all kinds of drugs, even levo-dopa (and herbals such as St John’s Wort or ephedra; for discussion, see part 2), may influence DAT imaging with [123I]FP-CIT SPECT. However, most centers use [123I]FP-CIT more frequently, or even exclusively, only for routine clinical studies. From data published and discussed in part 2, it is suggested that some medications or drugs of abuse are likely to influence the interpretation of [123I]FP-CIT SPECT images, which may lead to a misleading scan. Therefore, it is relevant prior to administration of [123I]FP-CIT to check the patient’s present and recent past medication record.
Cocaine and amphetamines including methylphenidate are the most clear examples of drugs (of abuse) that will influence the visual and quantitative analysis of [123I]FP-CIT SPECT scans. Therefore, to prevent a significant occupancy of the DAT, for routine clinical studies we recommend that the patient stop taking them at least five plasma clearance half-lives before the scan. This is a rather conservative approach because, in clinical trials, four plasma clearance half-lives is suggested to be an appropriate washout period for drugs [143]. However, the decision to withdraw any medication must always be made by the specialist in charge of the patient’s care, taking also into account the advantages and disadvantages of a withdrawal. Also, phentermine and ephedrines (as tablets; phentermine has previously been a prescription drugs but now is not generally available), modafinil, and some antidepressants (bupropion, radafaxine, and mazindol; mazindol has previously been a prescription drug but now is not generally available) are likely to influence the scan significantly, and should be stopped (if allowed; five half-lives). There are data to suggest that anticholinergics, and particularly benzatropine, may influence the visual and quantitative interpretation of FP-CIT SPECT scan significantly by reducing [123I]FP-CIT binding to the DAT due to its relatively high affinity for the DAT. Therefore, we recommend stopping benzatropine to prevent a misleading [123I]FP-CIT SPECT scan. Based on studies in rats and monkeys, other anticholinergics (such as scopolamine) might increase striatal [123I]FP-CIT binding ratios. For a visual analysis of FP-CIT scans, it is therefore not necessary to withdraw this medication. On the other hand, these drugs might lead to an overestimation of striatal FP-CIT binding ratios in humans. This increased striatal uptake may possibly affect the results of a quantitative analysis. There are also some data to suggest that opioids may influence the interpretation of FP-CIT SPECT scans. Therefore, fentanyl should not be used when [123I]FP-CIT is administered (at least five half-lives). For other opioids, the evidence that they may influence DAT imaging is not strong enough to advocate withdrawing them before a routine [123I]FP-CIT SPECT scan. Ketamine and PCP are sometimes used illicitly, and it is likely that these drugs will significantly influence the interpretation of the scan visually as well as quantitatively.
SSRIs and SNRI will significantly increase striatal FP-CIT binding ratios. However, because the effects will only be around 10%, we believe that, for an individual patient, such an effect will be too small to misinterpret an individual scan. Therefore, we do not recommend withdrawing this medication for routine clinical studies. Based on literature, antipsychotic medication and cholinesterase inhibitors should not be stopped prior to a routine [123I]FP-CIT SPECT scan. Finally, amantadine is frequently used in parkinsonian patients. Although animal studies showed effects on DAT, no human studies have been performed so far. Although it may be of value to test the influence of amantadine on [123I]FP-CIT SPECT in humans, until now there is no direct evidence which support an effect. Therefore, at this moment, we do not recommend stopping this medication.
Conclusion
In conclusion, based on published data, it is likely that several drugs (of abuse), e.g., amphetamines, will influence the visual interpretation and quantification of [123I]FP-CIT SPECT scans in routine clinical studies. Ideally, such medication should be stopped before the administration of the radiotracer. The decision to withdraw any medication must always be made by the specialist in charge of the patient’s care, taking into account the pros and cons of a withdrawal.
References
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 1973;20:415–55.
Kaufman MJ, Madras BK. Severe depletion of cocaine recognition sites associated with the dopamine transporter in Parkinson’s-diseased striatum. Synapse 1991;9:43–9.
Garnett ES, Firnau G, Nahmias C. Dopamine visualised in the basal ganglia of living man. Nature 1983;305:137–8.
Frey KA, Koeppe RA, Kilbourn MR, Vander Borght TM, Albin RL, Gilman S, et al. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Ann Neurol 1996;40:873–84.
Booij J, Tissingh G, Boer GJ, Speelman JD, Stoof JC, Janssen AG, et al. [123I]FP-CIT SPECT shows a pronounced decline of striatal dopamine transporter labelling in early and advanced Parkinson’s disease. J Neurol Neurosurg Psychiatry 1997;62:133–40.
Innis RB, Seibyl JP, Scanley BE, Laruelle M, Abi-Dargham A, Wallace E, et al. Single photon emission computed tomographic imaging demonstrates loss of striatal dopamine transporters in Parkinson disease. Proc Natl Acad Sci USA 1993;90:11965–9.
Halldin C, Erixon-Lindroth N, Pauli S, Chou YH, Okubo Y, Karlsson P, et al. [11C]PE2I: a highly selective radioligand for PET examination of the dopamine transporter in monkey and human brain. Eur J Nucl Med Mol Imaging 2003;30:1220–30.
Kung HF, Kim HJ, Kung MP, Meegalla SK, Plossl K, Lee HK. Imaging of dopamine transporters in humans with technetium-99m TRODAT-1. Eur J Nucl Med 1996;23:1527–30.
Rinne JO, Laihinen A, Nagren K, Ruottinen H, Ruotsalainen U, Rinne UK. PET examination of the monoamine transporter with [11C]β-CIT and [11C]β-CFT in early Parkinson’s disease. Synapse 1995;21:97–103.
Farde L, Ehrin E, Eriksson L, Greitz T, Hall H, Hedström CG, et al. Substituted benzamides as ligands for visualization of dopamine receptor binding in the human brain by positron emission tomography. Proc Natl Acad Sci USA 1985;82:3863–7.
Kung HF, Alavi A, Chang W, Kung MP, Keyes JW Jr, Velchik MG, et al. In vivo SPECT imaging of CNS D-2 dopamine receptors: initial studies with iodine-123-IBZM in humans. J Nucl Med 1990;31:573–9.
Mukherjee J, Christian BT, Dunigan KA, Shi B, Narayanan TK, Satter M, et al. Brain imaging of 18F-fallypride in normal volunteers: blood analysis, distribution, test–retest studies, and preliminary assessment of sensitivity to aging effects on dopamine D-2/D-3 receptors. Synapse 2002;46:170–88.
Booij J, Tissingh G, Winogrodzka A, van Royen EA. Imaging of the dopaminergic neurotransmission system using single-photon emission tomography and positron emission tomography in patients with parkinsonism [review]. Eur J Nucl Med 1999;26:171–82.
Marshall V, Grosset D. Role of dopamine transporter imaging in routine clinical practice [review]. Mov Disord 2003;18:1415–23.
Ponsen MM, Stoffers D, Booij J, van Eck-Smit BL, Wolters ECh, Berendse HW. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol 2004;56:173–81.
Stiasny-Kolster K, Doerr Y, Moller JC, Hoffken H, Behr TM, Oertel WH, et al. Combination of ‘idiopathic’ REM sleep behaviour disorder and olfactory dysfunction as possible indicator for alpha-synucleinopathy demonstrated by dopamine transporter FP-CIT-SPECT. Brain 2005;128:126–37.
O’Brien JT, Colloby S, Fenwick J, Williams ED, Firbank M, Burn D, et al. Dopamine transporter loss visualized with FP-CIT SPECT in the differential diagnosis of dementia with Lewy bodies. Arch Neurol 2004;61:919–25.
Walker Z, Costa DC, Walker RW, Shaw K, Gacinovic S, Stevens T, et al. Differentiation of dementia with Lewy bodies from Alzheimer’s disease using a dopaminergic presynaptic ligand. J Neurol Neurosurg Psychiatry 2002;73:134–40.
McKeith I, O’Brien J, Walker Z, Tatsch K, Booij J, Darcourt J, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol 2007;6:305–13.
Ishikawa T, Dhawan V, Kazumata K, Chaly T, Mandel F, Neumeyer J, et al. Comparative nigrostriatal dopaminergic imaging with iodine-123-beta-CIT-FP/SPECT and fluorine-18-FDOPA/PET. J Nucl Med 1996;37:1760–5.
Booij J, Busemann Sokole E, Stabin MG, Janssen AG, de Bruin K, van Royen EA. Human biodistribution and dosimetry of [123I]FP-CIT: a potent radioligand for imaging of dopamine transporters. Eur J Nucl Med 1998;25:24–30. Erratum in: Eur J Nucl Med 1998;25:458.
Booij J, Hemelaar TG, Speelman JD, de Bruin K, Janssen AG, van Royen EA. One-day protocol for imaging of the nigrostriatal dopaminergic pathway in Parkinson’s disease by [123I]FPCIT SPECT. J Nucl Med 1999;40:753–61.
Benamer TS, Patterson J, Grosset DG, Booij J, de Bruin K, van Royen E, et al. Accurate differentiation of parkinsonism and essential tremor using visual assessment of [123I]-FP-CIT SPECT imaging: the [123I]-FP-CIT study group. Mov Disord 2000;15:503–10.
Catafau AM, Tolosa E, [123I]FP-CIT Clinically Uncertain Parkinsonian Syndromes Study Group. Impact of dopamine transporter SPECT using 123I-Ioflupane on diagnosis and management of patients with clinically uncertain Parkinsonian syndromes. Mov Disord 2004;19:1175–82.
Varrone A, Pellecchia MT, Amboni M, Sansone V, Salvatore E, Ghezzi D, et al. Imaging of dopaminergic dysfunction with [123I]FP-CIT SPECT in early-onset parkin disease. Neurology 2004;63:2097–103.
Andringa G, Drukarch B, Bol JGJM, de Bruin K, Sorman K, Habraken JB, et al. Pinhole SPECT imaging of dopamine transporters correlates with dopamine transporter immunohistochemical analysis in the MPTP mouse model of Parkinson’s disease. Neuroimage 2005;26:1150–8.
Salvatore E, Varrone A, Sansone V, Nolano M, Bruni AC, De Rosa A, et al. Characterization of nigrostriatal dysfunction in spinocerebellar ataxia 17. Mov Disord 2006;21:872–5.
Spiegel J, Hellwig D, Mollers MO, Behnke S, Jost W, Fassbender K, et al. Transcranial sonography and [123I]FP-CIT SPECT disclose complementary aspects of Parkinson’s disease. Brain 2006;129:1188–93.
Lavalaye J, Linszen DH, Booij J, Dingemans PM, Reneman L, Habraken JB, et al. Dopamine transporter density in young patients with schizophrenia assessed with [123I]FP-CIT SPECT. Schizophr Res 2001;47:59–67.
Neumeyer JL, Wang S, Gao Y, Milius RA, Kula NS, Campbell A, et al. N-ω-fluoroalkyl analogs of (1R)-2β-carbomethoxy-3β-(4-iodophenyl)-tropane (β-CIT): radiotracers for positron emission tomography and single photon emission computed tomography imaging of dopamine transporters. J Med Chem 1994;37:1558–61.
Scheffel U, Lever JR, Abraham P, Parham KR, Mathews WB, Kopajtic T, et al. N-substituted phenyltropanes as in vivo binding ligands for rapid imaging studies of the dopamine transporter. Synapse 1997;25:345–9.
Laruelle M, Baldwin RM, Malison RT, Zea-Ponce Y, Zoghbi SS, al-Tikriti MS, et al. SPECT imaging of dopamine and serotonin transporters with [123I]β-CIT: pharmacological characterization of brain uptake in nonhuman primates. Synapse 1993;13:295–309.
Booij J, Andringa G, Rijks LJ, Vermeulen RJ, De Bruin K, Boer GJ, et al. [123I]FP-CIT binds to the dopamine transporter as assessed by biodistribution studies in rats and SPECT studies in MPTP-lesioned monkeys. Synapse 1997;27:183–90.
Vles JS, Feron FJ, Hendriksen JG, Jolles J, van Kroonenburgh MJ, Weber WE. Methylphenidate down-regulates the dopamine receptor and transporter system in children with attention deficit hyperkinetic disorder (ADHD). Neuropediatrics 2003;34:77–80.
Ginovart N, Wilson AA, Houle S, Kapur S. Amphetamine pretreatment induces a change in both D2-Receptor density and apparent affinity: a [11C]raclopride positron emission tomography study in cats. Biol Psychiatry 2004;55:1188–94.
Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, et al. Amphetamine-induced loss of human dopamine transporter activity: An internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci USA 2000;97:6850–5.
Byas-Smith M, Votaw J, Hua J, Voll R, Martarello L, Levey AI, et al. Phenylephrine and norepinephrine increase dopamine transporter ligand binding in striatum. Mol Imaging Biol 2003;5:217–26.
Kish SJ, Furukawa Y, Chang LJ, Tong J, Ginovart N, Wilson A, et al. Regional distribution of serotonin transporter protein in postmortem human brain. Is the cerebellum a SERT-free brain region? Nucl Med Biol 2005;32:123–8.
Reneman L, Booij J, Lavalaye J, de Bruin K, Reitsma JB, Gunning B, et al. Use of amphetamine by recreational users of ecstasy (MDMA) is associated with reduced striatal dopamine transporter densities: a [123I]β-CIT SPECT study—preliminary report. Psychopharmacology (Berl) 2002;159:335–40.
Kahlig KM, Galli A. Regulation of dopamine transporter function and plasma membrane expression by dopamine, amphetamine, and cocaine [review]. Eur J Pharmacol 2003;479:153–8.
Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci 1997;17:45–7.
Foster JD, Cervinski MA, Gorentla BK, Vaughan RA. Regulation of the dopamine transporter by phosphorylation. Handb Exp Pharmacol 2006;175:197–214.
Foster JD, Pananusorn B, Vaughan RA. Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J Biol Chem 2002;277:25178–86.
Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science 1999;285:763–6.
Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci 1999;19:7699–710.
Jayanthi LD, Ramamoorthy S. Regulation of monoamine transporters: influence of psychostimulants and therapeutic antidepressants [review]. AAPS J 2005;7:E728–38.
Gulley JM, Zahniser NR. Rapid regulation of dopamine transporter function by substrates, blockers and presynaptic receptor ligands. Eur J Pharmacol 2003;479:139–52.
Inaba T. Cocaine: pharmacokinetics and biotransformation in man. Can J Physiol Pharmacol 1989;67:1154–7.
Brandenberger H, Maes RAA, editors. Analytical toxicology for clinical, forensic, and pharmacuetical chemists. Berlin: Walter de Gruyter; 1977. p. 665–72.
Chaly T, Dhawan V, Kazumata K, Antonini A, Margouleff C, Dahl JR, et al. Radiosynthesis of [18F] N-3-fluoropropyl-2-β-carbomethoxy-3-β-(4-iodophenyl) nortropane and the first human study with positron emission tomography. Nucl Med Biol 1996;23:999–1004.
Bergström KA, Halldin C, Lundkvist C, Swahn C-G, Åkerman KK, Kuikka JT, et al. Characterization of C-11 or I-123 labelled β-CIT-FP and β-CIT-FE metabolism measured in monkey and human plasma. Identification of two labeled metabolites with HPLC. Hum Psychopharmacol 1996;11:483–90.
Kula NS, Baldessarini RJ, Tarazi FI, Fisser R, Wang S, Trometer J, et al. [3H]β-CIT: a radioligand for dopamine transporters in rat brain tissue. Eur J Pharmacol 1999;385:291–4.
Pellinen P, Honkakoski P, Stenback F. Cocaine N-demethylation and the metabolism-related hepatotoxicity can be prevented by cytochrome P450 3A inhibitors. Eur J Pharmacol 1994;270:35–43.
Yaqub M, Boellaard R, van Berckel BN, Ponsen MM, Lubberink M, Windhorst AD, et al. Quantification of dopamine transporter binding using [18F]FP-β-CIT and positron emission tomography. J Cereb Blood Flow Metab 2007;27:1397–406.
Lammertsma AA, Bench CJ, Price GW, Cremer JE, Luthra SK, Turton D, et al. Measurement of cerebral monoamine oxidase B activity using L-[11C]deprenyl and dynamic positron emission tomography. J Cereb Blood Flow Metab 1991;11:545–56.
Gunduz H, Wu H, Ashtari M, Bogerts B, Crandall D, Robinson DG, et al. Basal ganglia volumes in first-episode schizophrenia and healthy comparison subjects. Biol Psychiatry 2002;51:801–8.
Laruelle M, Abi-Dargham A, van Dyck C, Gil R, D’Souza DC, Krystal J, et al. Dopamine and serotonin transporters in patients with schizophrenia: an imaging study with [123I]β-CIT. Biol Psychiatry 2000;47:371–9.
Lavalaye J, Booij J, Reneman L, Habraken JB, van Royen EA. Effect of age and gender on dopamine transporter imaging with [123I]FP-CIT SPET in healthy volunteers. Eur J Nucl Med 2000;27:867–9.
Booij J, Habraken JB, Bergmans P, Tissingh G, Winogrodzka A, Wolters EC, et al. Imaging of dopamine transporters with iodine-123-FP-CIT SPECT in healthy controls and patients with Parkinson’s disease. J Nucl Med 1998;39:1879–84.
Qin Y, Ouyang Q, Pablo J, Mash DC. Cocaine abuse elevates alpha-synuclein and dopamine transporter levels in the human striatum. Neuroreport 2005;16:1489–93.
Malison RT, Best SE, van Dyck CH, McCance EF, Wallace EA, Laruelle M, et al. Elevated striatal dopamine transporters during acute cocaine abstinence as measured by [123I]β-CIT SPECT. Am J Psychiatry 1998;155:832–4.
Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, et al. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature 1997;386:827–30.
Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry 2001;158:377–82.
Chou YH, Huang WS, Su TP, Lu RB, Wan FJ, Fu YK. Dopamine transporters and cognitive function in methamphetamine abuser after a short abstinence: a SPECT study. Eur Neuropsychopharmacol 2007;17:46–52.
McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci 1998;180:8417–22.
Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, et al. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci 2001;21:9414–8.
Sekine Y, Iyo M, Ouchi Y, Matsunaga T, Tsukada H, Okada H, et al. Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am J Psychiatry 2001;158:1206–14.
Spencer TJ, Biederman J, Ciccone PE, Madras BK, Dougherty DD, Bonab AA, et al. PET study examining pharmacokinetics, detection and likeability, and dopamine transporter receptor occupancy of short- and long-acting oral methylphenidate. Am J Psychiatry 2006;163:387–95.
Gruber AJ, Pope HG. Ephedrine abuse among 36 female weightlifters. Am J Addict 1998;7:256–61.
Alexander M, Rothman RB, Baumann MH, Endres CJ, Brasic JR, Wong DF. Noradrenergic and dopaminergic effects of (+)-amphetamine-like stimulants in the baboon Papio anubis. Synapse 2005;56:94–9.
Wee S, Ordway GA, Woolverton WL. Reinforcing effect of pseudoephedrine isomers and the mechanism of action. Eur J Pharmacol 2004;493:117–25.
Rothman RB, Vu N, Partilla JS, Roth BL, Hufeisen SJ, Compton-Toth BA, et al. In vitro characterization of ephedrine-related stereoisomers at biogenic amine transporters and the receptorome reveals selective actions as norepinephrine transporter substrates. J Pharmacol Exp Ther 2003;307:138–45.
Rothman RB, Blough BE, Baumann MH. Dual dopamine-5-HT releasers: potential treatment agents for cocaine addiction [review]. Trends Pharmacol Sci 2006;27:612–8.
Mark EJ, Patalas ED, Chang HT, Evans RJ, Kessler SC. Fatal pulmonary hypertension associated with short-term use of fenfluramine and phentermine. N Engl J Med 1997;337:602–6. Erratum in: N Engl J Med 1997;337:1483.
Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular heart disease associated with fenfluramine–phentermine. N Engl J Med 1997;337:581–8. Erratum in: N Engl J Med 1997;337:1783.
Colman E. Anorectics on trial: a half century of federal regulation of prescription appetite suppressants. Ann Intern Med 2005;143:380–5.
Vaugeois JM, Bonnet JJ, Costentin J. In vivo labelling of the neuronal dopamine uptake complex in the mouse striatum by [3H]GBR 12783. Eur J Pharmacol 1992;210:77–84.
Giambalvo CT, Price LH. Effects of fenfluramine and antidepressants on protein kinase C activity in rat cortical synaptoneurosomes. Synapse 2003;50:212–22.
McCann UD, Seiden LS, Rubin LJ, Ricaurte GA. Brain serotonin neurotoxicity and primary pulmonary hypertension from fenfluramine and dexfenfluramine. A systematic review of the evidence. JAMA 1997;278:666–72.
Mignot E, Nishino S, Guilleminault C, Dement WC. Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep 1994;17:436–7.
Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergic role in stimulant-induced wakefulness. J Neurosci 2001;21:1787–94.
Madras BK, Xie Z, Lin Z, Jassen A, Panas H, Lynch L, et al. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther 2006;319:561–9.
Malison RT, McCance E, Carpenter LL, Baldwin RM, Seibyl JP, Price LH, et al. [123I]β-CIT SPECT imaging of dopamine transporter availability after mazindol administration in human cocaine addicts. Psychopharmacology 1998;137:321–5.
Argyelan M, Szabo Z, Kanyo B, Tanacs A, Kovacs Z, Janka Z, et al. Dopamine transporter availability in medication free and in bupropion treated depression: a 99mTc-TRODAT-1 SPECT study. J Affect Disord 2005;89:115–23.
Meyer JH, Goulding VS, Wilson AA, Hussey D, Christensen BK, Houle S. Bupropion occupancy of the dopamine transporter is low during clinical treatment. Psychopharmacology 2002;163:102–5.
Learned-Coughlin SM, Bergstrom M, Savitcheva I, Ascher J, Schmith VD, Langstrom B. In vivo activity of bupropion at the human dopamine transporter as measured by positron emission tomography. Biol Psychiatry 2003;54:800–5.
Kugaya A, Seneca NM, Snyder PJ, Williams SA, Malison RT, Baldwin RM, et al. Changes in human in vivo serotonin and dopamine transporter availabilities during chronic antidepressant administration. Neuropsychopharmacology 2003;28:413–20.
Reneman L, Booij J, Lavalaye J, de Bruin K, Reitsma JB, Gunning B, et al. Use of amphetamine by recreational users of ecstasy (MDMA) is associated with reduced striatal dopamine transporter densities: a [123I]β-CIT SPECT study—preliminary report. Psychopharmacology (Berl) 2002;159:335–40.
de Win MM, Habraken JB, Reneman L, van den Brink W, den Heeten GJ, Booij J. Validation of [123I]β-CIT SPECT to assess serotonin transporters in vivo in humans: a double-blind, placebo-controlled, crossover study with the selective serotonin reuptake inhibitor citalopram. Neuropsychopharmacology 2005;30:996–1005.
Booij J, de Jong J, de Bruin K, Knol RJJ, de Win MM, van Eck-Smit BLF. Quantification of striatal dopamine transporters with [123I]FP-CIT SPECT is influenced by the selective serotonin reuptake inhibitor paroxetine: a double-blind, placebo-controlled, crossover study in healthy controls. J Nucl Med 2007;48:359–66.
Owens MJ, Knight DL, Nemeroff CB. Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry 2001;50:345–50.
Tatsumi M, Groshan K, Blakely RD, Richelson E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 1997;340:249–58.
Scheffel U, Kim S, Cline EJ, Kuhar MJ. Occupancy of the serotonin transporter by fluoxetine, paroxetine, and sertraline: in vivo studies with [125I]RTI-55. Synapse 1994;16:263–8.
Inoue O, Kobayashi K, Hosoi R, Gee A. Opposing effects of clomipramine on [125I]RTI-55 and [3H]N-methylspiperone binding in mouse striatum: important role of other factors than endogenous dopamine? Synapse 1998;30:338–40.
Fujita M, Takatoku K, Matoba Y, Nishiura M, Kobayashi K, Inoue O, et al. Enhancement of [123I]β-CIT binding in the striatum with clomipramine: is there a serotonin–dopamine interaction? Eur J Nucl Med 1997;24:403–8.
Shang Y, Gibbs MA, Marek GJ, Stiger T, Burstein AH, Marek K, et al. Displacement of serotonin and dopamine transporters by venlafaxine extended release capsule at steady state: a [123I]2β-carbomethoxy-3β-(4-iodophenyl)-tropane single photon emission computed tomography imaging study. J Clin Psychopharmacol 2007;27:71–5.
Volkow ND, Wang GJ, Fowler JS, Learned-Coughlin S, Yang J, Logan J, et al. The slow and long-lasting blockade of dopamine transporters in human brain induced by the new antidepressant drug radafaxine predict poor reinforcing effects. Biol Psychiatry 2005;57:640–6.
Chen F, Lawrence AJ. The effects of antidepressant treatment on serotonergic and dopaminergic systems in Fawn-Hooded rats: a quantitative autoradiography study. Brain Res 2003;976:22–9.
Thibaut F, Bonnet JJ, Vaugeois JM, Costentin J. Pharmacological modifications of dopamine transmission do not influence the striatal in vivo binding of [3H]mazindol or [3H]cocaine in mice. Neurosci Lett 1996;205:145–8.
Tatsumi M, Jansen K, Blakely RD, Richelson E. Pharmacological profile of neuroleptics at human monoamine transporters. Eur J Pharmacol 1999;368:277–83.
Basta-Kaim A, Budziszewska B, Jaworska-Feil LM, Tetich M, Kubera M, Leskiewicz M, et al. Antipsychotic drugs inhibit the human corticotropin-releasing-hormone gene promoter activity in neuro-2A cells—an involvement of protein kinases. Neuropsychopharmacology 2006;31:853–65.
Mateos JJ, Lomena F, Parellada E, Mireia F, Fernandez-Egea E, Pavia J, et al. Lower striatal dopamine transporter binding in neuroleptic-naive schizophrenic patients is not related to antipsychotic treatment but it suggests an illness trait. Psychopharmacology (Berl) 2007;191:805–11.
Lavalaye J, Booij J, Reneman L, Habraken JB, van Royen EA. [123I]FP-CIT binding in rat brain after acute and sub-chronic administration of dopaminergic medication. Eur J Nucl Med 2000;27:346–9.
Emre M, Aarsland D, Albanese A, Byrne EJ, Deuschl G, De Deyn PP, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351:2509–18.
Thomas AJ, Burn DJ, Rowan EN, Littlewood E, Newby J, Cousins D, et al. A comparison of the efficacy of donepezil in Parkinson’s disease with dementia and dementia with Lewy bodies. Int J Geriatr Psychiatry 2005;20:938–44.
Tsukada H, Nishiyama S, Ohba H, Sato K, Harada N, Kakiuchi T. Cholinergic neuronal modulations affect striatal dopamine transporter activity: PET studies in the conscious monkey brain. Synapse 2001;42:193–5.
Taylor J-P, Colloby SJ, McKeith IG, Burn DJ, Williams D, Patterson J, et al. Cholinesterase inhibitor use does not significantly influence the ability of 123I-FP-CIT imaging to distinguish AD from DLB. J Neurol Neurosurg Psychiatry 2007;78:1069–71
Kilbourn MR, Kemmerer ES, Desmond TJ, Sherman PS, Frey KA. Differential effects of scopolamine on in vivo binding of dopamine transporter and vesicular monoamine transporter radioligands in rat brain. Exp Neurol 2004;188:387–90.
Lees 2005. Alternatives to levodopa in the initial treatment of early Parkinson’s disease. Drugs Aging 2005;22:731–740.
Madras BK, Fahey MA, Goulet M, Lin Z, Bendor J, Goodrich C, et al. Dopamine transporter (DAT) inhibitors alleviate specific parkinsonian deficits in monkeys: association with DAT occupancy in vivo. J Pharmacol Exp Ther 2006;319:570–85.
Lees AJ. Drugs for Parkinson’s disease [review]. J Neurol Neurosurg Psychiatry 2002;73:607–10.
Winogrodzka A, Booij J, Wolters ECh. Disease-related and drug-induced changes in dopamine transporter expression might undermine the reliability of imaging studies of disease progression in Parkinson’s disease [review]. Parkinsonism Relat Disord 2005;11:475–84.
Schillaci O, Pierantozzi M, Filippi L, Manni C, Brusa L, Danieli R, et al. The effect of levodopa therapy on dopamine transporter SPECT imaging with 123I-FP-CIT in patients with Parkinson’s disease. Eur J Nucl Med Mol Imaging 2005;32:1452–6.
Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med 2004;351:2498–508.
Innis RB, Marek KL, Sheff K, Zoghbi S, Castronuovo J, Feigin A, et al. Effect of treatment with L-dopa/carbidopa or L-selegiline on striatal dopamine transporter SPECT imaging with [123I]β-CIT. Mov Disord 1999;14:436–42.
Fowler JS, Volkow ND, Logan J, Franceschi D, Wang GJ, MacGregor R, et al. Evidence that L-deprenyl treatment for one week does not inhibit MAO A or the dopamine transporter in the human brain. Life Sci 2001;68:2759–68.
Heinonen EH, Anttila MI, Lammintausta RA. Pharmacokinetic aspects of l-deprenyl (selegiline) and its metabolites [review]. Clin Pharmacol Ther 1994;56:742–9.
Winogrodzka A, Bergmans P, Booij J, van Royen EA, Stoof JC, Wolters EC. [123I]β-CIT SPECT is a useful method for monitoring dopaminergic degeneration in early stage Parkinson’s disease. J Neurol Neurosurg Psychiatry 2003;74:294–8.
Ahlskog JE, Uitti RJ, O’Conor MK, Maraganore DM, Matsumoto JY, Stark KF, et al. The effect of dopamine agonist therapy on dopamine transporter imaging in Parkinson’s disease. Mov Disord 1999;14:940–6.
Guttman M, Stewart D, Hussey D, Wilson A, Houle S, Kish S. Influence of l-dopa and pramipexole on striatal dopamine transporter in early PD. Neurology 2001;56:1559–64.
Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I; Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 2004;291:317–24.
Page G, Peeters M, Maloteaux JM, Hermans E. Increased dopamine uptake in striatal synaptosomes after treatment of rats with amantadine. Eur J Pharmacol 2000;403:75–80.
Gordon I, Weizman R, Rehavi M. Modulatory effect of agents active in the presynaptic dopaminergic system on the striatal dopamine transporter. Eur J Pharmacol 1996;298:27–30.
Di Paolo T. Modulation of brain dopamine transmission by sex steroids. Rev Neurosci 1994;5:27–42.
Best SE, Sarrel PM, Malison RT, Laruelle M, Zoghbi SS, Baldwin RM, et al. Striatal dopamine transporter availability with [123I]β-CIT SPECT is unrelated to gender or menstrual cycle. Psychopharmacology (Berl) 2005;183:181–9.
Gardiner SA, Morrison MF, Mozley PD, Mozley LH, Brensinger C, Bilker W, et al. Pilot study on the effect of estrogen replacement therapy on brain dopamine transporter availability in healthy, postmenopausal women. Am J Geriatr Psychiatry 2004;12:621–30.
Collins SL, Gerdes RM, D’Addario C, Izenwasser S. Kappa opioid agonists alter dopamine markers and cocaine-stimulated locomotor activity. Behav Pharmacol 2001;12:237–45.
Bergström KA, Jolkkonen J, Kuikka JT. Fentanyl decreases β-CIT binding to the dopamine transporter. Synapse 1998;29:413–5.
Izenwasser S, Newman AH, Cox BM, Katz JL. The cocaine-like behavioral effects of meperidine are mediated by activity at the dopamine transporter. Eur J Pharmacol 1996;297:9–17.
Lomenzo SA, Izenwasser S, Gerdes RM, Katz JL, Kopajtic T, Trudell ML. Synthesis, dopamine and serotonin transporter binding affinities of novel analogues of meperidine. Bioorg Med Chem Lett 1999;9:3273–6.
Lomenzo SA, Rhoden JB, Izenwasser S, Wade D, Kopajtic T, Katz JL, et al. Synthesis and biological evaluation of meperidine analogues at monoamine transporters. J Med Chem 2005;48:1336–43.
Xiao ZW, Cao CY, Wang ZX, Li JX, Liao HY, Zhang XX. Changes of dopamine transporter function in striatum during acute morphine addiction and its abstinence in rhesus monkey. Chin Med J 2006;119:1802–7.
Simantov R. Chronic morphine alters dopamine transporter density in the rat brain: possible role in the mechanism of drug addiction. Neurosci Lett 1993;163:121–4.
Kish SJ, Kalasinsky KS, Derkach P, Schmunk GA, Guttman M, Ang L, et al. Striatal dopaminergic and serotonergic markers in human heroin users. Neuropsychopharmacology 2001;24:561–7.
Beekman F, van der Have F. The pinhole: gateway to ultra-high-resolution three-dimensional radionuclide imaging. Eur J Nucl Med Mol Imaging 2007;34:151–61.
Cherry SR. The 2006 Henry N. Wagner lecture: of mice and men (and positrons)—advances in PET imaging technology. J Nucl Med 2006;47:1735–45.
Tsukada H, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Harada N. Ketamine alters the availability of striatal dopamine transporter as measured by [11C]β-CFT and [11C]β-CIT-FE in the monkey brain. Synapse 2001;42:273–80.
Harada N, Ohba H, Fukumoto D, Kakiuchi T, Tsukada H. Potential of [18F]β-CFT-FE (2β-carbomethoxy-3β-(4-fluorophenyl)-8-(2-[18F]fluoroethyl)nortropane) as a dopamine transporter ligand: a PET study in the conscious monkey brain. Synapse 2004;54:37–45.
Byas-Smith MG, Li J, Szlam F, Eaton DC, Votaw JR, Denson DD. Isoflurane induces dopamine transporter trafficking into the cell cytoplasm. Synapse 2004;53:68–73.
Nishimura M, Sato K. Ketamine stereoselectively inhibits rat dopamine transporter. Neurosci Lett 1999;274:131–4.
Riley SC, James C, Gregory D, Dingle H, Cadger M. Patterns of recreational drug use at dance events in Edinburgh, Scotland. Addiction 2001;96:1035–47.
Schiffer WK, Logan J, Dewey SL. Positron emission tomography studies of potential mechanisms underlying phencyclidine-induced alterations in striatal dopamine. Neuropsychopharmacology 2003;28:2192–8.
Greenblatt DJ. Basic pharmacokinetic principles and their application to psychotropic drugs. J Clin Psychiatry 1993;54:S8–13.
Acknowledgements
The authors would like to thank Remco JJ Knol (MD) for the production of Fig. 1. The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Booij, J., Kemp, P. Dopamine transporter imaging with [123I]FP-CIT SPECT: potential effects of drugs. Eur J Nucl Med Mol Imaging 35, 424–438 (2008). https://doi.org/10.1007/s00259-007-0621-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-007-0621-0