
Abstract
3′-Deoxy-3′-18F-fluorothymidine (FLT) was developed in 1998 by Shields and co-workers because monitoring of treatment response would be facilitated by imaging agents able to provide measures of tissue and tumour proliferation. Since then, FLT metabolism has been clarified in more detail in cell culture and experimental animal tumour models and also in clinical studies. Recently, FLT has increasingly been used for the assessment of response to anticancer treatment, mainly in tumour xenograft SCID mouse models; in contrast, clinical data are scarce. In this article we briefly summarise the intermediary metabolism of FLT and its application as an anticancer treatment response probe. The potential value and limitations of FLT as a highly promising proliferation imaging probe and its use for monitoring of treatment response are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Assessment of tumour response to cytoreductive treatment is an essential component of the management of patients with malignant disease in clinical practice and serves as an objective end point in clinical trials. The response of solid tumours to anticancer agents and/or radiotherapy is commonly assessed by radiological imaging techniques. The WHO first established response assessment criteria in 1979 to standardise the recording and reporting of response assessment of solid tumours [1]. In 1994, several international health organisations formulated the revised RECIST criteria for solid tumour response assessment [2] (Table 1).
As shown in Table 1, the employed criteria are mostly anatomically defined. Recently, functional and molecular imaging MR and PET techniques have been developed and introduced into clinical practice [3–5]. PET has demonstrated the ability to detect changes in tumour metabolism prior to changes in the actual size of the tumour, and to predict outcome earlier and more precisely [6–8]. The performance of 18F-fluorodeoxyglucose (FDG) PET in evaluating tumour response is reviewed in detail in this issue by Lammertsma, Mikhaeel and Stroobants. Here, we shall briefly summarise the intermediary metabolism of 3′-deoxy-3′-18F-fluorothymidine (FLT) and review its application as an anticancer treatment response probe.
Metabolism of FLT
The concept of using tumour uptake of FLT as a surrogate marker for tumour proliferation was developed because early experiments using 11C-thymidine PET to measure proliferation were not successful. Rapid degradation of 11C-thymidine by thymidine phosphorylase (EC 2.4.2.4), present in blood, liver, spleen and tumour tissue, resulted in a high blood radioactivity background owing to the many 11C-labelled metabolites [9]. For the same reason, iodine-labelled desoxyuridine was inappropriate for measuring proliferation in vivo [10, 11]. FLT is more stable against thymidine phosphorylase-mediated degradation and was thus developed and tested as a potential proliferation marker for PET [12].
After intravenous injection, FLT distributes rapidly in the extracellular fluid and is transported from there into the cytosol via nucleoside transporters, probably mostly via the equilibrative nucleoside transporter ENT1 and in some tissues, such as the intestines, also via the Na+-dependent concentrative nucleoside transporter CNT1. The transmembranous flux of FLT, however, has not yet been well characterised. It has been shown that ENT1 expression is upregulated severalfold in various tumour cell lines [13, 14], fostering the concept of using radiolabelled nucleosides for imaging and functional characterisation of malignant tumours.
Within the cytosol, FLT is phosphorylated by thymidine kinase 1 (TK1) (EC 2.7.1.21). TK1 is a cell cycle regulated enzyme with a severalfold increased expression during the S phase [15–17]. In many malignant tumours, TK1 is constitutively upregulated. FLT is a selective substrate for TK1 [18]. In contrast, thymidine and other 2-arabinofuranosyl-nucleosides such as FIAU [5-iodo-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)uracil] are also phosphorylated by the mitochondrial enzyme thymidine kinase 2 (TK2) (EC 2.7.1.21), which is expressed independently of the cell cycle, leading to significant cardiac and liver toxicity when they are used as antiviral agents with therapeutic intent [19].
In proliferating cells, FLT is metabolised through the anabolic arm of the thymidine salvage pathway. This pathway has an important role in balancing intracellular nucleotide pools. It operates by using futile phosphorylation/dephosphorylation cycles to adjust intracellular nucleotide pools, derived from de novo synthesis and nucleoside influx from extracellular fluid and phosphorylated by TK1 [20]. The catabolic arm of these futile cycles involves a set of nucleotidases (e-NT, dNT-1, dNT-2, cN-I, cN-II), dephosphorylating excess deoxynucleotides. These nucleotidases have different substrate specificities and tissue distributions [21–24]. Nucleosides may efflux from the cytosol through facilitated, nucleoside transporter-mediated transport. Additional outward transport may be accomplished through a nucleoside transporter efflux pump [25].
FLT monophosphate (FLTMP) is phosphorylated by thymidylate kinase (TMPK) to FLT diphosphate (FLTDP) and subsequently to FLT triphosphate (FLTTP) by nucleotide diphosphate kinase (NDPK) [26–29]. Phosphorylation of thymidine monophosphate by TMPK is considered irreversible, whereas non-specific phosphorylation of nucleotide diphosphates by NDPK is probably reversible [20, 30–32]. NDPK is a ubiquitous, S phase-regulated kinase [32], overexpressed in some tumours [33].
It has been shown that in A549 lung adenocarcinoma cells, TMPK is the rate-limiting enzyme for FLT metabolism within the anabolic arm of the thymidine salvage pathway [25]. FLTMP NT-mediated dephosphorylation and subsequent FLT efflux was about 50% of total FLT initially accumulated within this tumour cell line [25].
Due to the 3′ substitution with 18F, FLT cannot be incorporated into the growing DNA chain but leads to DNA chain termination [26, 27]. Whether FLT, bound to the end of the growing DNA chain, is a substrate for DNA repair enzymes, for instance nucleoside excision repair enzymes, has, to our knowledge, not yet been examined.
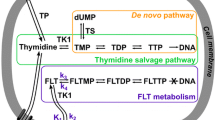
In summary, FLT enters the cell mainly through ENT1-mediated facilitated transport. Intracellular radioactivity accumulation results exclusively from formation of phosphorylated FLT mono-, di- and triphosphate nucleotides in competition with dNT-1-mediated dephosphorylation and FLT efflux [25]. The kinetics of this process is dominated by the expression and activities of TK1, dNT-1 and TMPK, the rate-limiting step in the net conversion of FLT to FLTTP being the activity of TMPK [25] (Fig. 1).

Metabolic pathway of FLT according to Grierson et al. [25]
Treatment evaluation in preclinical studies
The suitability of FLT for assessing anticancer treatment response has been assessed in several cell culture and tumour xenograft mouse models (Table 2, modified from ref. [34]). In most cell culture and virtually all tumour xenograft mouse models, a decrease in cellular FLT uptake was reported (Table 2, Fig. 2). Interestingly, increased FLT uptake was observed in several cell culture models when the thymidine synthesis pathway was blocked with 5-fluorouracil (5-FU) and/or methotrexate [35], suggesting that the salvage pathway might have been upregulated to adjust thymidine demands to actual supply. It might be hypothesised that this rather fragile metabolic situation can only be maintained within a post-therapeutic time window, when thymidine flux through the salvage pathway can really meet the demands for DNA synthesis and cell viability is not compromised. In those studies comparing treatment-associated changes in FLT and FDG uptake, an earlier and more pronounced post-treatment decrease in cellular or tumour uptake of FLT was observed. In addition, the temporary increase in FDG uptake after radiation therapy was not observed with FLT.

Treatment response to 5-FU evaluated with FLT and a small animal PET scanner in a fibrosarcoma SCID mouse xenograft model [54]
Clinical studies
Only very limited clinical data are available on treatment response assessment with FLT alone or in comparison to FDG. As reviewed by Beets et al. [36] and re-assessed in several recent studies, tumour uptake of FLT has been compared with that of FDG in several clinical tumour entities [37–41]. In general, FLT uptake is considerably lower than that of FDG, and in the case of non-small cell lung cancer (NSCLC), FLT uptake has been found in only 50% of FDG-positive nodal metastases (Buck, submitted). Therefore it is currently unclear whether the sensitivity of FLT imaging of primary assessable tumour lesions according to RECIST criteria is sufficient for reliable assessment of tumour response. In addition, physiological high uptake in haematopoietic bone marrow and the liver will certainly render assessment of small bone/bone marrow and liver metastases difficult.
Buck et al. and Vesselle et al. were the first to demonstrate a clear-cut correlation of tumour FLT uptake, measured with PET, and tumour proliferation rate, measured with Ki67 immunostaining of proliferating cells in NSCLC resection or biopsy specimens [38, 42]. These findings were recently confirmed by Muzi et al. [43] and Yap et al. [44].
In follicular lymphoma, a close correlation of FLT uptake with proliferation rate was recently observed by our group (Buck et al., submitted, Fig. 3). The results of this study indicated that progression of indolent to aggressive lymphoma might be assessed with FLT PET (Buck et al., submitted). Also in brain tumours, tumour FLT uptake correlated with Ki67-measured tumour proliferation rate [45]. In contrast, Smyczek-Gargya et al. did not observe a significant correlation of tumour FLT uptake and Ki67-assessed tumour proliferation rate in patients with breast cancer [46]. In summary, also in a clinical setting, most researchers have observed a correlation of tumour proliferation rate and tumour FLT uptake, measured with PET. These observations clearly support the concept of using FLT for the evaluation of cancer treatment.

a FLT PET whole-body scan of a patient with an aggressive NHL. Supra- and infradiaphragmatic nodal disease with intense FLT accumulation and a high proliferation rate shown by immunohistology (Ki67 staining) of an excised node. b Close correlation of FLT uptake with Ki67-positive cells in patients with NHL (Buck et al., submitted)
A literature search has identified only one published study on the assessment of treatment response with FLT in breast cancer [47]. In a very small sample of 14 patients with newly diagnosed primary or metastatic breast cancer, patients had baseline FDG and FLT scans, scans with each tracer at 2 weeks after termination of the first treatment cycle (chemotherapy or antihormonal therapy), and final scans with each tracer after the end of treatment or after 1 year if treatment had not yet been terminated. Treatment-related changes in FLT and FDG uptake were generally concordant [47]. The authors found that alteration in the blood concentration of the tumour marker Ca 27.29 was more strongly associated with treatment-induced changes in tumour FLT uptake than with changes in FDG uptake. The authors felt that FLT PET performed 2 weeks after the end of the first treatment course was useful in predicting long-term efficacy of the applied treatment [47]. Although the results of this study are encouraging, it is clear that a much more comprehensive understanding of the major determinants of tumour FLT metabolism in a clinical setting is necessary before a solid judgement can be made on the ability of FLT to measure anticancer treatment response.
For example, key enzymes of thymidine synthesis such as thymidylate synthase can be upregulated severalfold during 5-FU or methotrexate treatment [48, 49]. Treatment-induced changes in tumour protein expression profiles are considered a major mechanism of resistance to radiochemotherapy [50]. It is conceivable that nucleoside transporters and enzymes of thymidine metabolism display similar treatment-induced changes in their expression pattern. Therefore, anticancer treatment-related changes in the function and expression of key proteins and enzymes of thymidine metabolism need to be known before quantitative measures of the flux of FLT through the thymidine salvage pathway can be used as a reliable treatment response parameter.
In addition, to the best of our knowledge, the uptake properties of FLT in individual tumour cells in vivo have not been examined as yet. Therefore, the intrinsic assumption underlying the use of FLT as a treatment response probe, namely that all tumour cells are equally and homogeneously labelled by FLT in vivo, remains to be proven. Also, the minimum tumour cell mass that can be imaged with FLT in vivo in a clinical setting, which will define the sensitivity of this novel imaging test, needs to be determined.
In summary, it is clear that we are just beginning the process of defining the role of FLT as an anticancer treatment response probe. Nevertheless, its ability to specifically label an important pathway of tumour cell metabolism renders FLT a highly promising and potentially important novel radiopharmaceutical. Rapid progress in our knowledge of basic treatment-related changes in tumour FLT metabolism and in the definition of its clinical utility is foreseeable in the near future.
References
Hricak H, Akin O, Bradbury MS, Lieberman L, Schwartz LH, Larson SM. Advanced imaging methods: functional and metabolic imaging. In: DeVita VT, Hellman S, Rosenberg SA, editors. Cancer: principles and practice of oncology. 7 ed. Philadelphia: Lippincott Williams & Wilkins; 2005. p. 589–720
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92:205–16
Reske SN, Kotzerke J. FDG-PET for clinical use. Results of the 3rd German Interdisciplinary Consensus Conference, “Onko-PET III”, 21 July and 19 September 2000. Eur J Nucl Med 2001;28:1707–23
Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer 2002;2:683–93
Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci U S A 2000;97:9226–33
Jerusalem G, Beguin Y, Fassotte MF, Belhocine T, Hustinx R, Rigo P, et al. Early detection of relapse by whole-body positron emission tomography in the follow-up of patients with Hodgkin’s disease. Ann Oncol 2003;14:123–30
Weber WA, Ott K. Imaging of esophageal and gastric cancer. Semin Oncol 2004;31:530–41
Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N Engl J Med 2006;354:496–507
Shields AF, Mankoff D, Graham MM, Zheng M, Kozawa SM, Link JM, et al. Analysis of 2-carbon-11-thymidine blood metabolites in PET imaging. J Nucl Med 1996;37:290–6
Tjuvajev JG, Macapinlac HA, Daghighian F, Scott AM, Ginos JZ, Finn RD, et al. Imaging of brain tumor proliferative activity with iodine-131-iododeoxyuridine. J Nucl Med 1994;35:1407–17
Blasberg RG, Roelcke U, Weinreich R, Beattie B, von Ammon K, Yonekawa Y, et al. Imaging brain tumor proliferative activity with [124I]iododeoxyuridine. Cancer Res 2000;60:624–35
Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med 1998;4:1334–6
Plagemann PG, Richey DP, Zylka JM, Erbe J. Thymidine transport by Novikoff rat hepatoma cells synchronized by double hydroxyurea treatment. Exp Cell Res 1974;83:303–10
Hopwood LE, Dewey WC, Hejny W. Transport of thymidine during the cell cycle in mitotically synchronized CHO cells. Exp Cell Res 1975;96:425–9
Munch-Petersen B, Cloos L, Jensen HK, Tyrsted G. Human thymidine kinase 1. Regulation in normal and malignant cells. Adv Enzyme Regul 1995;35:69–89
Sherley JL, Kelly TJ. Regulation of human thymidine kinase during the cell cycle. J Biol Chem 1988;263:8350–8
Eriksson S, Munch-Petersen B, Johansson K, Eklund H. Structure and function of cellular deoxyribonucleoside kinases. Cell Mol Life Sci 2002;59:1327–46
Munch-Petersen B, Cloos L, Tyrsted G, Eriksson S. Diverging substrate specificity of pure human thymidine kinases 1 and 2 against antiviral dideoxynucleosides. J Biol Chem 1991;266:9032–8
Wang J, Eriksson S. Phosphorylation of the anti-hepatitis B nucleoside analog 1-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)-5-Iodouracil (FIAU) by human cytosolic and mitochondrial thymidine kinase and implications for cytotoxity. Antimicrob Agents Chemother 1996;40:1555–7
Kornberg A, Baker TA. Biosynthesis in DNA precursors. DNA replication. 2nd ed. New York: W.H. Freeman; 1992. p. 53–100
Skladanowski AC, Hoffmann C, Krass J, Jastorff B, Makarewicz W. Structure-activity relationship of cytoplasmic 5′-nucleotidase substrate sites. Biochem J 1996;314 Pt 3:1001–7
Garvey EP, Lowen GT, Almond MR. Nucleotide and nucleoside analogues as inhibitors of cytosolic 5′-nucleotidase I from heart. Biochemistry 1998;37:9043–51
Gazziola C, Ferraro P, Moras M, Reichard P, Bianchi V. Cytosolic high K(m) 5′-nucleotidase and 5′(3′)-deoxyribonucleotidase in substrate cycles involved in nucleotide metabolism. J Biol Chem 2001;276:6185–90
Mansson E, Flordal E, Liliemark J, Spasokoukotskaja T, Elford H, Lagercrantz S, et al. Down-regulation of deoxycytidine kinase in human leukemic cell lines resistant to cladribine and clofarabine and increased ribonucleotide reductase activity contributes to fludarabine resistance. Biochem Pharmacol 2003;65:237–47
Grierson JR, Schwartz JL, Muzi M, Jordan R, Krohn KA. Metabolism of 3′-deoxy-3′-[F-18]fluorothymidine in proliferating A549 cells: validations for positron emission tomography. Nucl Med Biol 2004;31:829–37
Kong XB, Zhu QY, Vidal PM, Watanabe KA, Polsky B, Armstrong D, et al. Comparisons of anti-human immunodeficiency virus activities, cellular transport, and plasma and intracellular pharmacokinetics of 3′-fluoro-3′-deoxythymidine and 3′-azido-3′-deoxythymidine. Antimicrob Agents Chemother 1992;36:808–18
Sundseth R, Joyner S, Moore J, Dornsife R, Dev I. The anti-human immunodeficiency virus agent 3′-fluorothymidine induces DNA damage and apoptosis in human lymphoblastoid cells. Antimicrob Agents Chemother 1996;40:331–5
Langen P, Etzold G, Hintsche R, Kowollik G. 3′-Deoxy-3′-fluorothymidine, a new selective inhibitor of DNA-synthesis. Acta Biol Med Ger 1969;23:759–66
Matthes E, Lehmann C, Scholz D, Rosenthal HA, Langen P. Phosphorylation, anti-HIV activity and cytotoxicity of 3′-fluorothymidine. Biochem Biophys Res Commun 1988;153:825–31
Stryer L. Biochemistry. 4 ed. New York: W.H. Freeman; 1995
Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol Ther 2000;87:189–98
Caligo MA, Cipollini G, Fiore L, Calvo S, Basolo F, Collecchi P, et al. NM23 gene expression correlates with cell growth rate and S-phase. Int J Cancer 1995;60:837–42
Lacombe M-L, Milon L, Munier A. The human Nm23/nucleoside diphosphate kinases. J Bioenerg Biomembr 2000;32:247–58
Been LB, Suurmeijer AJH, Cobben DCP, Jager PL, Hoekstra HJ, Elsinga PH. [18F]FLT-PET in oncology: current status and opportunities. Eur J Nucl Med Mol Imaging 2004;31:1659–72
Dittmann H, Dohmen BM, Kehlbach R, Bartusek G, Pritzkow M, Sarbia M, et al. Early changes in [18F]FLT uptake after chemotherapy: an experimental study. Eur J Nucl Med Mol Imaging 2002;29:1462–9
Beets G, Penninckx F, Schiepers C, Filez L, Mortelmans L, Kerremans R, et al. Clinical value of whole-body positron emission tomography with [18F]fluorodeoxyglucose in recurrent colorectal cancer. Br J Surg 1994;81:1666–70
Buck AK, Halter G, Schirrmeister H, Kotzerke J, Wurziger I, Glatting G, et al. Imaging proliferation in lung tumors with PET:18F-FLT versus18F-FDG. J Nucl Med 2003;44:1426–31
Vesselle H, Grierson J, Muzi M, Pugsley JM, Schmidt RA, Rabinowitz P, et al. In vivo validation of 3′deoxy-3-[18F]fluorothymidine ([18F]FLT) as a proliferation imaging tracer in humans: correlation of [18F]FLT uptake by positron emission tomography with Ki-67 immunohistochemistry and flow cytometry in human lung tumors. Clin Cancer Res 2002;8:3315–23
Cobben DC, Elsinga PH, Hoekstra HJ, Suurmeijer AJ, Vaalburg W, Maas B, et al. Is18F-3′-fluoro-3′-deoxy-L-thymidine useful for the staging and restaging of non-small cell lung cancer? J Nucl Med 2004;45:1677–82
Cobben DC, Laan B, Maas B, Vaalburg W, Suurmeijer AJ, Hoekstra HJ, et al.18F-FLT PET for visualization of laryngeal cancer: comparison with18F-FDG PET. J Nucl Med 2004;45:226–31
van Westreenen HL, Cobben DC, Jager PL, van Dullemen HM, Wesseling J, Elsinga PH, et al. Comparison of18F-FLT PET and18F-FDG PET in esophageal cancer. J Nucl Med 2005;46:400–4
Buck AK, Schirrmeister H, Hetzel M, Von Der Heide M, Halter G, Glatting G, et al. 3-deoxy-3-[18F]fluorothymidine-positron emission tomography for noninvasive assessment of proliferation in pulmonary nodules. Cancer Res 2002;62:3331–4
Muzi M, Vesselle H, Grierson JR, Mankoff DA, Schmidt RA, Peterson L, et al. Kinetic analysis of 3′-deoxy-3′-fluorothymidine PET studies: validation studies in patients with lung cancer. J Nucl Med 2005;46:274–82
Yap CS, Czernin J, Fishbein MC, Cameron RB, Schiepers C, Phelps ME, et al. Evaluation of thoracic tumors with 18F-fluorothymidine and18F-fluorodeoxyglucose-positron emission tomography. Chest 2006;129:393–401
Chen W, Cloughesy T, Kamdar N, Satyamurthy N, Bergsneider M, Liau L, et al. Imaging proliferation in brain tumors with18F-FLT PET: comparison with18F-FDG. J Nucl Med 2005;46:945–52
Smyczek-Gargya B, Fersis N, Dittmann H, Vogel U, Reischl G, Machulla HJ, et al. PET with [18F]fluorothymidine for imaging of primary breast cancer: a pilot study. Eur J Nucl Med Mol Imaging 2004;31:720–4
Pio BS, Park CK, Pietras R, Hsueh WA, Satyamurthy N, Pegram MD, et al. Usefulness of 3′-[F-18]fluoro-3′-deoxythymidine with positron emission tomography in predicting breast cancer response to therapy. Mol Imaging Biol 2006;8:36–42
Jakob C, Liersch T, Meyer W, Baretton GB, Hausler P, Schwabe W, et al. Immunohistochemical analysis of thymidylate synthase, thymidine phosphorylase, and dihydropyrimidine dehydrogenase in rectal cancer (cUICC II/III): correlation with histopathologic tumor regression after 5-fluorouracil-based long-term neoadjuvant chemoradiotherapy. Am J Surg Pathol 2005;29:1304–9
Ma T, Zhu ZG, Ji YB, Zhang Y, Yu YY, Liu BY, et al. Correlation of thymidylate synthase, thymidine phosphorylase and dihydropyrimidine dehydrogenase with sensitivity of gastrointestinal cancer cells to 5-fluorouracil and 5-fluoro-2′-deoxyuridine. World J Gastroenterol 2004;10:172–6
Rosenwald A, Staudt LM. Clinical translation of gene expression profiling in lymphomas and leukemias. Semin Oncol 2002;29:258–63
Waldherr C, Mellinghoff IK, Tran C, Halpern BS, Rozengurt N, Safaei A, et al. Monitoring antiproliferative responses to kinase inhibitor therapy in mice with 3′-deoxy-3′-18F-fluorothymidine PET. J Nucl Med 2005;46:114–20
Sugiyama M, Sakahara H, Sato K, Harada N, Fukumoto D, Kakiuchi T, et al. Evaluation of 3′-deoxy-3′-18F-fluorothymidine for monitoring tumor response to radiotherapy and photodynamic therapy in mice. J Nucl Med 2004;45:1754–8
Leyton J, Latigo JR, Perumal M, Dhaliwal H, He Q, Aboagye EO. Early detection of tumor response to chemotherapy by 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography: the effect of cisplatin on a fibrosarcoma tumor model in vivo. Cancer Res 2005;65:4202–10
Barthel H, Cleij MC, Collingridge DR, Hutchinson OC, Osman S, He Q, et al. 3′-deoxy-3′-[18F]fluorothymidine as a new marker for monitoring tumor response to antiproliferative therapy in vivo with positron emission tomography. Cancer Res 2003;63:3791–8
Oyama N, Ponde DE, Dence C, Kim J, Tai YC, Welch MJ. Monitoring of therapy in androgen-dependent prostate tumor model by measuring tumor proliferation. J Nucl Med 2005;45:519–25
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reske, S.N., Deisenhofer, S. Is 3′-deoxy-3′-18F-fluorothymidine a better marker for tumour response than 18F-fluorodeoxyglucose?. Eur J Nucl Med Mol Imaging 33 (Suppl 1), 38–43 (2006). https://doi.org/10.1007/s00259-006-0134-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-006-0134-2