
Abstract
The aim of this study was to evaluate the properties of the D-amino acid isomers O-18F-fluoromethyl tyrosine (18F-FMT), O-18F-fluoroethyl tyrosine (18F-FET) and O-18F-fluoropropyl tyrosine (18F-FPT) as tumour-detecting agents with PET in comparison with the corresponding L-isomers. L- or D-18F-FMT, 18F-FET or 18F-FPT, prepared by 18F-fluoromethylation, 18F-fluoroethylation or 18F-fluoropropylation of L- and D-tyrosine, was intravenously injected into BALB/cA Jcl-nu mice bearing HeLa tumour cells. At 5, 15, 30 and 60 min post intravenous administration, the uptake of each compound in normal abdominal organs and xenotransplanted HeLa cells was determined using the tissue dissection method. Metabolic stability analyses of these compounds in the plasma were performed with the thin-layer chromatography method. In the plasma fraction, although L- and D-isomers of 18F-FMT, 18F-FET and 18F-FPT provided comparable metabolic stability, D-isomers of these labelled compounds revealed a faster elimination rate than their L-isomers, with a higher peak uptake in the blood and kidney 5 min post administration. Compared with natural amino acid ligands, such as L-11C-methionine, the uptake of L-isomers of these labelled compounds was relatively low and stable in the abdominal organs, while D-isomers revealed much lower and faster clearance rates compared with the corresponding L-isomers. Among the abdominal organs, the pancreas showed relatively high uptake of all the labelled compounds used here, and the uptake of D-isomers was much lower than that of the L-isomers. Although tumour uptake levels of D-isomers of 18F-FMT, 18F-FET and 18F-FPT were almost 95%, 43% and 39% of the uptake levels of each of the L-isomers 60 min post administration, the tumour-to-blood ratios of these D-isomers were 181%, 137% and 101% of the ratios of the corresponding L-isomers. D-isomers of 18F-FMT and 18F-FET indicated improved tumour-to-liver ratios compared with the corresponding L-isomers, and D-18F-FPT showed the highest tumour-to-pancreas ratio among all the other compounds assayed here. These results suggest that D-isomers of 18F-fluoroalkyl tyrosine analogues are potential tracers for tumour imaging with PET.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The in vivo regional protein synthesis rate in tissue has been quantitatively determined by the combined use of positron emission tomography (PET) and positron-labelled natural amino acids or unnatural amino acid analogues. Because of the low amino acid utilisation rate in the cortical normal tissues, radiolabelled amino acids and their analogues are considered to be useful, especially for tumour detection in the brain. Their use in tumour detection is primarily based on an increased uptake of amino acids, which is assumed to reflect an enhanced amino acid metabolism and protein synthesis. Natural and unnatural artificial labelled amino acids, 1-11C-methionine [1, 2], methyl-11C-methionine (11C-MET) [2–4], 1-11C-tyrosine [5, 6], β-11C-tyrosine (11C-TYR) [7], 1-11C-leucine (11C-LEU) [8], 1-11C-phenylalanine (11C-PHE) [9, 10], 4-18F-fluoro-L-phenylalanine [11] and 2-18F-fluoro-L-tyrosine [12, 13] have been proposed (for review see 14). Among these labelled amino acids, L-11C-MET is widely used for tumour imaging with PET. Since it has been reported that L-11C-MET is incorporated not only into protein fractions via the conversion into amino-acyl-tRNA, but also into non-protein materials, such as lipids and RNA, via the transmethylation process via S-adenosyl-L-methionine [2], the value of this tracer is considered to be as a marker of methionine transport into tissue in vivo for imaging tumour tissues with PET [14]. Influenced by this working hypothesis, several artificial amino acid tracers, O-11C-methyl-L-tyrosine (CMT) [15, 16], O-18F-fluoromethyl-L-tyrosine (FMT) [15, 16], O-18F-fluoroethyl-L-tyrosine (FET) [17, 18] and O-18F-fluoropropyl-L-tyrosine (FPT) [19, 20], have been synthesised and evaluated for their potential to be better tumour imaging agents. These radiolabelled compounds were found to behave as unnatural amino acids after intravenous injection, exhibiting relatively low accumulation in normal peripheral tissues (low tissue-to-blood ratio), except for the pancreas. No significant elevation of radioactivity with time was found in normal peripheral tissues, suggesting a remarkably low incorporation ratio into the protein fraction. These compounds also showed relatively slow blood clearance and relatively high uptake into tumour tissue (high tumour-to-blood ratio). Furthermore, Ishiwata et al. [21] demonstrated that other artificial amino acids, L-[ethyl-11C]ethionine (L-[11C]ETH) and L-[propyl-11C]propionine (L-[11C]PRO), S-ethyl- and S-propyl-substituted analogues of L-[11C]MET, are also potent tumour imaging agents.
Since L-isomers of amino acids are commonly used in mammalian cells in nature, D-isomers are considered to be unnatural amino acids, and radiolabelled D-amino acids are therefore expected to be good tumour imaging agents. However, the reported results have been somewhat controversial. It has been reported that D-isomers of some radiolabelled amino acids, such as 14C-MET [22] and 14C-LEU [23], show higher uptake into xenotransplanted tumour cells than the corresponding L-isomers. We also recently demonstrated in tumour-bearing mice that D-isomers of 11C-MET, 11C-CMT and 18F-FMT could be better tumour imaging agents than their L-isomers [24]. Schober et al. [25] showed no enantiomeric selective transport of L- and D-11C-MET into a malignant glioma. In contrast, Bergström et al. [26] demonstrated that uptake of D-11C-MET into brain tumours was lower than that of L-11C-MET. Furthermore, D-18F-FET showed negligible accumulation in human colon carcinoma cells in an in vitro assay [18], and also in the mouse brain in vivo [17], compared with its L-isomer, suggesting the stereospecificity of amino acid transport across the cell membrane and blood-brain barrier (BBB).
The aim of this study was to evaluate the potential of D-isomers of 18F-FMT and its o-fluoroalkyl substituted tyrosine analogues, 18F-FET and 18F-FPT, as tumour imaging agents in comparison with their corresponding L-isomers in tumour-bearing mice.
Materials and methods
Animals
Female BALB/cA Jcl-nu mice (6 weeks old) were obtained from Japan Clea (Tokyo, Japan). Mice were housed five per cage under standard laboratory conditions at 25°C and 50% humidity with a 12-h light/dark cycle (light on at 6.00 a.m., light-off at 6.00 p.m.) throughout the experimental period. They were allowed free access to food and water. Mice were inoculated subcutaneously with 5×106 of JCRB9004 HeLa cells (doubling time =4.4 days) when 7 weeks old, maintained for 2 weeks after xenotransplantation with monitoring of growth rates, and subjected to the experiments at 9 weeks of age. The tumour volume and weight used in the present study were 631.5±102.1 mm3 and 517±129 mg, respectively. Mice were maintained and handled in accordance with the recommendations of the US National Institutes of Health and also the guidelines of the Central Research Laboratory, Hamamatsu Photonics.
Chemicals
Ethylene glycol di-p-tosylate, 1,3-propanediol-di-p-tosylate, dimethylsulphoxide (anhydrous) (DMSO), N,N-dimethylformamide (anhydrous) (DMF), acetonitrile (anhydrous) and D-tyrosine were purchased from Sigma-Aldrich Japan (Tokyo, Japan). 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo- [8,8,8]hexacosane (K[2,2,2]) and K2CO3·1.5H2O were from Merck (Darmstadt, Germany). Anion exchange resin AG1-X8 (OH- form, 100-200 mesh) was from Bio-Rad Laboratories (Hercules, CA, USA). L-Tyrosine and dibromomethane were obtained from Nacalai Tesque (Kyoto, Japan). All other reagents were of analytical grade.
Syntheses of labelled compounds
Positron-emitting fluorine-18 (18F) was produced by 18O(p, n)18F nuclear reaction using the cyclotron (HM-18, Sumitomo Heavy Industry, Osaka, Japan) at Hamamatsu Photonics PET centre.L- and D-18F-FMT, 18F-FET and 18F-FPT were synthesised by reactions of 18F-fluoromethyl bromide, 18F-fluoroethyl tosylate or 18F-fluoropropyl tosylate with the corresponding L- and D-tyrosine. After irradiation, 18F-fluoride was recovered from target by He flow. Then, the recovered 18F-fluoride solution was trapped by ion-exchange resin (AG1-X8, Bio-Rad) and eluted from resin by 0.5 ml of 40 mM K2CO3 solution. To this fluoride solution, 2 ml of K[2,2,2] solution (containing 15 mg of K[2,2,2] in CH3CN) was added. Water was removed by azeotropic distillation at 110°C under He flow (400 ml/min) for 5 min. To the residue, the addition of acetonitrile (1 ml) and azeotropic distillation was repeated twice. Then, the residue was dried under reduced pressure for 1 min and under He flow (50 ml/min) for 1 min. Cooling the residue to room temperature gave the activated 18F-fluoride/K[2,2,2] complex.To synthesise the no-carrier-added 2-18F-fluoromethyl bromide (18F-FMBr), 0.1 ml of dibromomethane in 1 ml of CH3CN was added to 18F-fluoride/K[2,2,2] complex described above, and it was reacted at 110°C for 4 min. 18F-FMBr was distilled into precursor solution through in-line connected Sep-Pak silica cartridges. Then, 18F-FMBr fraction was collected into precursor solution of L- or D-tyrosine (2 mg/ml) in DMSO (0.3 ml) at −10°C for subsequent alkylation. After reacting for 5 min at room temperature, the reaction mixture was diluted with 1.0 ml of water and applied to high-performance liquid chromatography (HPLC): column, YMC-Pack ODS-A column (10 mm inner diameter × 250 mm length, particle size 5 μm, YMC Co. Ltd., Kyoto, Japan); mobile phase, CH3CN/50 mM CH3CO2H/50 mM CH3CO2NH4 (8/46/46, v/v/v); and flow rate, 5 ml/min. The 18F-FMT fraction was evaporated to dryness. The residue was dissolved in physiological saline and sterilised by membrane filtration.
For the synthesis of no-carrier-added 2-18F-fluoroethyl tosylate (18F-FEOTs), 5 mg of ethyleneglycol-di-tosylate in 1.2 ml of CH3CN was added to 18F-fluoride/K[2,2,2] complex described above. Fluorination was conducted at 110°C for 5 min. After cooling the reaction mixture, 4 ml of n-hexane/ether (=3/1) was added and applied to a Sep-Pak silica cartridge. Sep-Pak was washed with 8 ml of n-hexane/ether (=3/1). The combined eluate was concentrated under He flow at 80°C to give partially purified 18F-FEOTs. To prepare the no-carrier-added 2-18F-fluoropropyl tosylate (18F-FPOTs), it was prepared with the same procedure as that for 18F-FEOTs from 5 mg of 1,3-propanediol-di-p-tosylate. For 18F-fluoroethylation or fluoropropylation, 18F-FEOTs or 18F-FPOTs was added to the precursor solution of L- or D-tyrosine (2 mg/ml) in DMSO (0.3 ml) followed by heating at 90°C for 15 min; the reaction mixture was then cooled to room temperature. Thereafter, the mixture was passed through a silica Sep-Pak cartridge and C-18 Sep-Pak cartridge, and eluted with anhydrous ether (10 ml) and water (1 ml) to remove unreacted substrates. 18F-FET or 18F-FPT was eluted with 4 ml of 0.15 mol/l phosphate buffer, diluted with 2 ml of saline and applied to the same HPLC system as described for 18F-FMT preparation. 18F-FET or 18F-FPT fraction was evaporated, and the residue was dissolved in physiological saline and sterilised by membrane filtration.
Enantiomeric purity was analysed on a CHIOBIOTIC T column (4.6 mm×250 mm, Tokyo Kasei Kogyo, Tokyo, Japan). The elution solution was ethanol:water=1:1, and the flow rate was 1 ml/min.
Tissue distribution assay
Five MBq of a radiolabelled compound was injected into mice via the tail vein. The animals were sacrificed by decapitation under halothane anaesthesia 5, 15, 30 and 60 min after the injection. Samples of blood, heart, lung, liver, pancreas, kidney, spleen, small intestine, gut and tumour were rapidly removed, and the weight and radioactivity were measured using a gamma counter (Aloka ARC-2000, Tokyo, Japan). For the metabolite analysis, blood samples were centrifuged to separate the plasma, weighed and the radioactivity measured. Then, methanol was added to some of the plasma samples (sample/methanol=1/1), centrifuged and the obtained supernatants developed on thin-layer chromatography (TLC) plates (AL SIL G/UV, Whatman, Kent, UK) using a mobile phase of n-butanol/acetic acid/PBS (pH=7.4)=4/1/2. At each sampling time point for analysis, the ratio of radioactivity in the unmetabolised fraction to that in total plasma (metabolised plus unmetabolised) was determined using a phosphoimaging plate (BAS-1500 MAC, Tokyo, Japan). Standard uptake values (SUVs) were calculated as the radioactivity in tissue divided by the ratio of total injected radioactivity and body weight. Ratios of the SUVs of the organs and blood were calculated in a further analysis.
Statistical analysis
Results are expressed as means±SD. Comparisons between conditions were carried out using the unpaired, two-tailed Student’s t test. A probability level of less than 5% (p<0.05) was considered to indicate statistical significance.
Results
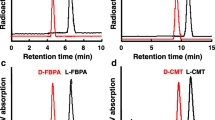
In order to evaluate the D-isomer of amino aid analogues as tumour-detecting agents, the quality control of enantiomeric purity was a critical factor. The retention times determined on the analytical HPLC were as follows: L- and D-FMT, 5.16 and 6.74 min; L- and D-FET, 4.85 and 6.66 min; L- and D-FPT, 5.48 and 7.05 min, respectively. As shown in Fig. 1, the results revealed that the enantiomeric purities of these six radiolabelled compounds exceeded 98%.

HPLC analyses of enantiomeric purities of L- and D-isomers of 18F-FMT (a), 18F-FET (b) and 18F-FPT (c). Enantiomeric purity of each compound was analysed on a CHIOBIOTIC T column using an elution solution of ethanol:water=1:1 at a flow rate of 1 ml/min
The tissue distribution and kinetics of L- and D-isomers of 18F-FMT, 18F-FET and 18F-FPT in mice are summarised in Tables 1, 2 and 3, expressed as the SUV. The kinetic data showed that the longer 18F-fluoroalkyl chain length induced the higher blood levels in the early phase after the injection with either the L- or D-isomers. In addition, all D-isomers of these labelled compounds revealed higher peak levels 5 min post injection, followed by a faster elimination rate than their L-isomers. As shown in Fig. 2, metabolic analysis in the plasma demonstrated that these compounds were generally stable, and that the stability of L-18F-fluoroalkyl tyrosine analogues was not significantly affected by changing the alkyl chain length. Of interest, the D-isomers had a slightly higher stability in the plasma than the corresponding L-isomers (Fig. 2a–c).

Metabolic analyses of L- and D-isomers of 18F-FMT (a), 18F-FET (b) and 18F-FPT (c) in the plasma of HeLa-bearing mice. At each time point after the injection, plasma samples were prepared, and the obtained samples were developed on TLC plates using a mobile phase of n-butanol/acetic acid/PBS (pH 7.4)=4/1/2. The ratio of radioactivity in the unmetabolised fraction to that in total plasma (metabolised plus unmetabolised) was determined using a phosphoimaging plate
In parallel to the blood kinetics, the uptake of all D-isomers in the kidney was tentatively much higher 5 min post injection and lower 30 min and later post injection than the uptake of the corresponding L-isomers (Tables 1, 2 and 3). Compared with the L-isomers, the uptake of the corresponding D-isomers in the liver was also higher 5 min post injection and lower thereafter (Tables 1, 2 and 3). In the pancreas and muscle, L-18F-FMT and L-18F-FET revealed almost similar uptake levels, and L-18F-FPT was slightly, but significantly lower than the former two compounds throughout the study. In contrast, the D-isomers indicated that the substitution by a longer 18F-fluoroalkyl chain length resulted in a lower uptake in the pancreas, but no significant difference in the muscle (data not shown). Compared with natural amino acid ligands, such as 11C-methionine, the uptake of L-isomers of 18F-FMT, 18F-FET and 18F-FPT was relatively low and stable in the abdominal organs, while D-isomers revealed much lower uptake and faster clearance rates compared with the corresponding L-isomers. Among the abdominal organs, the pancreas showed a markedly high uptake of all the L-isomers of the labelled compounds used here; however, the uptake of D-isomers was much lower than that of the L-isomers (Tables 1, 2 and 3). In addition, D-isomers with the longer 18F-fluoroalkyl chain length provided a lower pancreatic uptake level; in particular, the D-18F-FPT uptake in the pancreas was quite similar to that in the kidney and less than 60% of tumour uptake 60 min post injection (Table 3).
As shown in Tables 1, 2 and 3, L- and D-isomers of 18F-FMT, 18F-FET and 18F-FPT were gradually accumulated in the tumour tissue at least up to 45 min post injection. The tumour uptake of L-isomers of 18F-FMT, 18F-FET and 18F-FPT was not affected by the 18F-fluoroalkyl chain length, while the uptake of D-isomers was markedly lowered by the increase in the chain length (Tables 1, 2 and 3). Although the tumour uptake levels, as expressed by the SUV, of D-isomers of 18F-FMT, 18F-FET and 18F-FPT were almost 95%, 43% and 39%, respectively, of the tumour uptake levels of each of the L-isomers 60 min post administration (Tables 1, 2 and 3), the tumour-to-blood ratios of these D-isomers were 181%, 137% and 101%, respectively, of the ratios of the corresponding L-isomers (Fig. 3). To assess the contrast with the abdominal organs, the tumour-to-liver and tumour-to-pancreas ratios were compared among these labelled compounds. All L- and D-isomers used here provided tumour-to-liver ratios of more than 1.0 at 60 min post injection, and the tumour-to-liver ratios of the D-isomers were 198%, 132% and 123%, respectively, of the ratios of the corresponding L-isomers (Fig. 4a–c). Although the tumour-to-pancreas ratios of the D-isomers were also 250%, 195% and 288%, respectively, of those of the corresponding L-isomers, only D-18F-FPT provided a tumour-to-pancreas ratio of markedly more than 1.0 at 60 min post injection (Fig. 4d–f).

Time courses of tumour-to-blood ratios of L- and D-isomers of 18F-FMT (a), 18F-FET (b) and 18F-FPT (c) in HeLa-bearing mice. Ratios of SUV of tumour against SUV of blood were determined, then plotted against the time point after injection. *p<0.05 vs the corresponding L-isomer

Time courses of tumour-to-liver (a–c) and tumour-to-pancreas (d–f) ratios of L- and D-isomers of 18F-FMT (a, d), 18F-FET (b, e) and 18F-FPT (c, f) in HeLa-bearing mice. Ratios of SUV of tumour against SUV of liver or pancreas were determined, and then plotted against the time point after injection. *p<0.05 vs the corresponding L-isomer
Discussion
The present study investigated the capability of D-isomers of three 18F-labelled unnatural amino acids, 18F-FMT, 18F-FET and 18F-FPT, to image tumours by comparison with the corresponding L-isomers, which were previously reported not to be utilised in protein synthesis [16–18, 20].
Evaluation of enantiomeric purity with a stereospecific analytical column could certainly confirm that the L- and D-18F-FMT, 18F-FET and 18F-FPT applied in the present study had enantiomeric purities of more than 98%. The enantiomeric purity of these compounds was a critical issue, because the distribution and kinetics of these D-isomers in normal abdominal organs was revealed to be significantly different from those of the corresponding L-isomers. If a labelled compound with low enantiomeric purity were to be injected, one would expect the results of tumour grading and therapeutic efficacy to be affected because differences in the composition ratio of L- and D-isomers are responsible for differences in uptake values in the same tumour tissue.
It is well known that the high tumour uptake of amino acids mainly reflects the increased amino acid metabolism of tumour cells, including increased active transport and an increased protein synthesis rate. In general, natural L-amino acids are distributed into the free amino acid pool of tumour tissue across the plasma membranes, and some are actively utilised for protein synthesis in normal organs, such as the pancreas [27, 28]. The high uptake of labelled amino acids in normal organs impairs their use as tumour imaging agents because of the resultant lower tumour-to-normal tissue ratios. Our previous study demonstrated that the utilisation not only of L-11C-MET but also of D-11C-MET for protein synthesis was significantly reduced by pre-treatment with cycloheximide, a protein synthesis inhibitor [24]. These results may explain why D-11C-MET was not as good a tumour imaging agent as expected; D-11C-MET might be partly converted to α-keto-γ-methiolbutyrate by D-amino acid oxidase followed by transamination to its L-isomer [29, 30], resulting in high uptake in the plasma and normal abdominal organs [24]. In contrast, the uptake of D-18F-FMT into the protein fraction was found not to be affected by cycloheximide [24]. In addition, although pre-treatment with cycloheximide slightly decreased the incorporation of L-18F-FMT into the protein fraction, this did not hamper its role as a tumour imaging agent because the baseline utilisation level was almost negligible as the precursor amino acid of protein synthesis [24]. Several previous results have suggested that amino acid transport is the dominant uptake mechanism, rather than protein synthesis [14, 16, 31]. Although a cycloheximide study has not been performed, as for the utilisation in protein synthesis, L- and D-isomers of 18F-FET and 18F-FPT are expected to have similar properties to L- and D-18F-FMT. Thus, the L- and D-18F-FET and 18F-FPT taken up in the normal cells are rapidly excreted from the acid-soluble fraction into the urine owing to the lesser protein utilisation.
In the case of natural amino acids like L-11C-MET, the substitutions of the alkyl moiety from methyl to ethyl (L-11C-ethionine; L-11C-ETH) and propyl (L-11C-propionine; L-11C-PRO) groups resulted in different uptake patterns in the normal organs, as well as tumour tissue, as reported previously [21]. Thus, L-11C-ETH provided lower uptake into normal organs than L-11C-MET owing to its much lower utilisation in protein synthesis, and L-11C-PRO was not incorporated into the acid-precipitable fractions, showing similar properties to other artificial amino acids [14]. These results demonstrated that the increase in the alkyl chain length of L-11C-MET shifted its properties from those of a natural to those of an unnatural artificial amino acid. Since previous evaluations of tumour uptake of L-18F-FMT [16], 18F-FET [17, 18] and 18F-FPT [20] were conducted using different kinds of mice and inoculated tumours, it has been difficult to judge which labelled compound is most suitable as a tumour imaging agent. The present study firstly evaluated the effects of substitution of the 18F-fluoromethyl moiety of L-18F-FMT by 18F-fluoroethyl and 18F-fluoropropyl moieties on the distribution and kinetics in the same kind of experimental animal (BALB/cA Jcl-nu mice) bearing the same kind of tumour (HeLa cells). As a result, the substitutions did not induce such drastic changes in the distribution and kinetic pattern in normal organs of mice, or in HeLa tumour cells, as were observed in L-11C-MET series. No significant differences were detected in the tumour uptake of L-isomers of 18F-FMT, 18F-FET and 18F-FPT. In contrast, it was only observed in L-isomers that the longer length of 18F-fluoroalkyl chain provided a slight tendency for a gradual increase in radioactivity levels in the blood and liver, especially at 5 min post injection. Overall, the shorter 18F-fluoroalkyl chain length tyrosine provided a better tumour-to-blood ratio in L-isomer series.
In contrast to the L-isomer series, the tumour uptake of the D-isomer series of 18F-fluoroalkyl tyrosine was markedly lowered by the increase in the 18F-fluoroalkyl chain length, while the longer 18F-fluoroalkyl chain length provided slightly higher levels in the blood and liver. As a result, D-18F-FMT demonstrated the highest tumour-to-blood and tumour-to-liver ratios among the six labelled tyrosine analogues assayed here. It was observed that although the uptake of D-18F-FET in tumour was lower than that of L-18F-FET, D-18F-FET provided sufficient tumour uptake, as well as an adequate tumour-to-blood ratio, for tumour imaging. This result, however, seems to be at odds with previous reports that D-18F-FET showed negligible accumulation in human colon carcinoma cells in an in vitro assay [18] or in the mouse brain in vivo [17]. The discrepancy might be attributable to the difference between the in vitro and in vivo assay systems and also the difference in the tissues assayed. In fact, our preliminary in vitro data indicated a much lower uptake of D-18F-FET than its L-isomer in the HeLa cells used here for the assessment of uptake in the in vivo evaluation (unpublished data).
It was of interest that the D-isomers of 18F-FMT, 18F-FET and 18F-FPT showed a drastic decrease in pancreatic uptake in comparison with the corresponding L-isomers. It should be noted that only D-18F-FPT, which showed the lowest uptake levels among these six compounds at the later period post injection (ca. 45 min and beyond), provided a tumour-to-pancreas ratio of markedly more than 1.0. Although additional clinical investigations are necessary, this result suggests that, despite showing the lowest absolute uptake level in tumour tissue, D-18F-FPT might have potential for PET imaging of pancreatic tumours owing to better contrast between tumour and normal tissue.
Taking the previous and present results in conjunction, it needs to be considered whether synergic effects of the enantiomeric purity and the alkyl chain length on some currently unknown transporter system on the cell membrane might explain the differences in tissue uptake in vivo. When L-isomers were applied, there were no significant differences in uptake of FMT, FET and FPT as determined by SUVs, revealing that the transport system provides the tolerance against the length of alkyl chains including the lipophilicity of, at least, the artificial amino acid analogues. In contrast, the uptake values of D-isomers were critically affected by the alkyl chain length, suggesting that the structural changes induced synergistically by enantiomeric purity and the alkyl chain length might be beyond the tolerance range of the transport system. Against this background, a more precise analysis of the uptake mechanism might be needed.
In conclusion, the present results suggest that D-isomers of 18F-fluoroalkyl tyrosine analogues are potential tracers for tumour imaging with PET. All D-isomers assayed here had improved properties as tumour-detecting agents compared with the corresponding L-isomers. It should be emphasised especially that, among the six labelled L- and D-isomeric tyrosine analogues, D-18F-FMT provided the highest tumour-to-blood and tumour-to-liver ratios, and also that only D-18F-FPT provided a tumour-to-pancreas ratio of markedly more than 1.0 in mice bearing inoculated HeLa tumour cells.
References
Bolster JW, Vaalburg W, Elsinga PH, Wijnberg H, Woldring MG. Synthesis of DL-[1-11C]methionine. Appl Radiat Isot 1986;37:1069–1070
Ishiwata K, Vaalburg W, Elsinga PH, Paans AMJ, Woldring MG. Comparison of L-[1-11C]methionine and L-methyl-[11C]methionine for measuring in vivo protein synthesis rates with PET. J Nucl Med 1988;29:1419–1427
Comar D, Cartron JC, Maziere M, Marazano C. Labeling and metabolism of methionine-methyl-11C. Eur J Nucl Med 1976;1:11–14
Långström B, Antoni G, Gulberg P, Halldin C, Malmborg P, Någren K, et al. Synthesis of L and D-[methyl-11C]methionine. J Nucl Med 1987;28:1037–1040
Halldin C, Schoeps KO, Stone-Elander S, Wiesel FA. The Bucherer-Strecker synthesis of D- and L-[1-11C]tyrosine and the in vivo study of L-[1-11C]tyrosine in human brain using positron emission tomography. Eur J Nucl Med 1987;13:288–291
Ishiwata K, Vaalburg W, Elsinga PH, Paans AMJ, Woldring MG. Metabolic studies with L-[11C]tyrosine for the investigation of a kinetic model to measure protein synthesis rates with PET. J Nucl Med 1988;29:524–529
Bjurling P, Watanabe Y, Oka S, Nagasawa T, Yamada H, Långström B. Multienzymatic synthesis of β-11C-labelled L-tyrosine and L-DOPA. Acta Chem Scand 1990;44:183–188
Hawkins RA, Huang SC, Barrio JR, Keen RE, Feng D, Maziotta JC, et al. Estimation of local cerebral protein synthesis rates with L-[1-11C]leucine and PE: methods, model and results in animals and humans. J Cereb Blood Flow Metab 1989;9:446–460
Casey DL, Digenis GA, Wesner DA, Washburn LC, Chaney JE, Hayes RL, et al. Preparation and preliminary tissue studies of optically active D- and L-[11C]phenylalanine. Int J Appl Radiat Isot 1981;32:1291–1300
Barrio JR, Keen RE, Chugani H, Ackerman R, Chugani D, Phelps ME. L-[11C]Phenylalanine for the determination of cerebral protein synthesis rates in man with positron emission tomography. J Nucl Med 1983;24:70
Lemaire C, Guillaume M, Christiaens L, Palmer AJ, Cantineau R. A new route for the synthesis of [18F]fluoroaromatic substituted amino acids: no carrier acids L-p-[18F]fluorophenylalanine. Int J Radiat Appl 1987;38:1033–1038
Coenen HH, Kling P, Stöcklin G. Cerebral metabolism of L-[2-18F]fluorotyrosine, a new PET tracer of protein synthesis. J Nucl Med 1989;30:1367–1372
Wienhard K, Herholz K, Coenen HH, Rudolf J, Kling P, Stocklin G, et al. Increased amino acid transport into brain tumors measured by PET of L-(2-18F)fluorotyrosine. J Nucl Med 1991;32:1338–1346
Vaalburg W, Coenen HH, Crouzel C, Elsinga PH, Långström B, Lemaire C, et al. Amino acids for the measurement of protein synthesis in vivo by PET. Nucl Med Biol 1992;19:227–237
Iwata R, Furumoto S, Pascali C, Bogni A, Ishiwata K. Radiosynthesis of O-[11C]methyl-L-tyrosine and O-[18F]fluoromethyl-L-tyrosine as potential PET tracer for imaging amino acid transport. J Label Compd Radioparm 2003;46:555–566
Ishiwata K, Kawamura K, Wang WF, Furumoto S, Kubota K, Pascli C, et al. Evaluation of O-[11C]methyl-L-tyrosine and O-[18F]fluoromethyl-L-tyrosine as tumor imaging tracers by PET. Nucl Med Biol 2004;31:191–198
Wester HJ, Herz M, Weber W, Heiss P, Senekowitsch-Schmidtke R, Schwaiger M, et al. Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-tyrosine for tumor imaging. J Nucl Med 1999;40:205–212
Heiss P, Mayer S, Herz M, Wester HJ, Schwaiger M, Senekowitsch-Schmidtke R. Investigation of transport mechanism and uptake kinetics of O-(2-[18F]fluoroethyl)-L-tyrosine in vitro and in vivo. J Nucl Med 1999;40:1367–1373
Tang G, Tang X, Wang M, Luo L, Gan M. Fully automated synthesis of O-(3-[18F]fluoropropyl)-L-tyrosine by direct nucleophilic exchange on a quaternary 4-aminopyridium resin. Appl Radiat Isot 2003;58:685–689
Tang G, Wang M, Tang X, Luo L, Gan M. Synthesis and evaluation of O-(3-[18F]fluoropropyl)-L-tyrosine as an oncologic PET tracer. Nucl Med Biol 2003;30:733–739
Ishiwata K, Kasahara C, Hatano K, Ishii S, Senda M. Carbon-11 labeled ethionine and propionine as tumor detecting agents. Ann Nucl Med 1997;11:115–122
Takeda A, Goto R, Tamemasa O, Chaney JE, Digenis GA. Biological evaluation of radiolabeled D-methionine as a parent compound in potential nuclear imaging. Radioisotopes 1984;33:213–217
Tamemasa O, Goto R, Takeda A, Maruo K. High uptake of 14C-labeled D-amino acids by various tumors. Gann 1982;73:147–152
Tsukada H, Kengo S, Fukumoto D, Nishiyama S, Harada N, Kakiuchi T. Evaluation of D-isomers of O-11C-methyl tyrosine and O-18F-fluoromethyl tyrosine, as tumor imaging agents in tumor-bearing mice: a comparison study with L- and D-11C-methionine. J Nucl Med 2006;47:679–688
Schober O, Duden C, Meyer GJ, Muller JA, Hundeshagen H. Nonselective transport of [11C-methyl]-L- and D-methionine into a malignant glioma. Eur J Nucl Med 1987;13:103–105
Bergström M, Lundqvist H, Ericson K, Lilia A, Johanström P, Långström B, et al. Comparison of the accumulation kinetics of L-(methyl-11C)-methionine and D-(methyl-11C)-methionine in brain tumors studies with positron emission tomography. Acta Radiol 1987;28:225–229
Goto R, Tezuka M, Tamemasa O. Incorporation of L-, D-, and DL-amino acids into the pancreas of mice. Chem Pharm Bull 1977;25:1574–1581
Tamemasa O, Digenis GA, Tezuka M, Takeda A, Chaney LE, Goto R. Differential radioactivity uptake from 14C-labeled D- and L-leucine by the pancreas of animals pretreated with pancreatitis-causing agents. Chem Pharm Bull 1982;30:2521–2528
Oxender DL, Christensen HN. Distinct mediating system for the transport of natural amino acids by the Ehrich cell. J Biol Chem 1963;238:3686–3699
Christensen HN. On the development of amino acid transport system. Fed Proc 1973;32:19–28
Kubota K, Yamada K, Fukuda H, Endo S, Ito M, Abe Y, et al. Tumor detection with carbon-11-labelled amino acids. Eur J Nucl Med 1984;9:136–140
Acknowledgements
We gratefully acknowledge the excellent technical assistance provided by Shingo Nishiyama and Norihiro Harada in the synthesis of the labelled compounds. This study was supported in part by Research and Development of Technology for Measuring Vital Function Merged with Optical Technology, Research and Development Project Aimed at Economic Revitalisation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsukada, H., Sato, K., Fukumoto, D. et al. Evaluation of D-isomers of O-18F-fluoromethyl, O-18F-fluoroethyl and O-18F-fluoropropyl tyrosine as tumour imaging agents in mice. Eur J Nucl Med Mol Imaging 33, 1017–1024 (2006). https://doi.org/10.1007/s00259-006-0076-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-006-0076-8