
Abstract
This paper deals with a field experiment, combining the push–pull and tracer tests, conducted under natural gradient conditions at the international Oslo airport. The studied aquifer, showing very complex hydrogeological settings, has been contaminated by a jet fuel spill. The tracer solutes—bromide, toluene, o-xylene, 1,2,4-trimethylbenzene, 1,3,5-trimethylbenzene and naphthalene, have been injected into the plume. Their migration and changes in concentration of the electron acceptors and metabolic by-products have been monitored. Fast removal of both the non-reactive tracer as well as the aromatic organics has been observed. The tracer pulse could only be detected 2 m downgradient from the injection points. At this point, toluene and o-xylene have been completely removed, however, trimethylbenzenes and naphthalene have been detected. Their depletion, based on calculations of available electron acceptors, can, to a large extent, be accounted for intrinsic biodegradation, with Fe(III) and sulphate reductions as the major controlling processes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Groundwater contamination by the hazardous, toxic organic compounds has recently become an urgent issue of a great social and environmental concern. Common use of the hydrocarbons for fuelling cars, plains, etc. caused development of the storing and distributing facilities. Handling the petroleum products has often resulted in the accidental spills. The water soluble components of the petroleum products, like BTEX (benzene, toluene, ethylbenzene, xylenes) and naphthalene have been found to be rather mobile in the soils and groundwater, causing deterioration of the natural environment (Bennett et al. 1993; Eganhouse et al. 1993; Harvey 1998; Wiedemeier et al. 1999). Therefore, it is very important to understand the behaviour of the organic pollutants in the groundwater environment and to examine the controlling factors and processes of their migration and removal, like advective–dispersive transport, sorption, retardation, volatilisation and biodegradation (Fetter 1992; Fitts 2002; McAllister and Chiang 1994; Schwartz and Zhang 2003; Wiedemeier et al. 1999). The processes leading to a decrease of concentration and mass of the organic contaminants and/or to their complete removal have been termed natural attenuation and have lately become of a major interest (Chapelle 1999, 2001; Wiedemeier et al. 1999). The main process controlling degradation of the organics, often called intrinsic biodegradation (Wiedemeier et al. 1999), largely depends on availability of the electron acceptors which in turn, to a great extent, depends on the groundwater chemistry, flow direction and velocity as well as on geochemisry of aquifer solids (Alfnes et al. 2003a, b; Soevik et al. 2002).
The tracer and push–pull tests have been successfully applied to examine behaviour of organic pollutants upon transport within both pristine and contaminated aquifers. Such experiments, which involve monitoring of the injected non-reactive and reactive solutes, allow determination of the major factors governing the overall process of organic solutes degradation (Barker et al. 1987; Mackay et al. 1994, 1986; Schroth et al. 2001; Thierrin et al. 1995).
This paper deals with the push–pull and tracer tests conducted at the firefighting training site of the Oslo international airport. The concentrations of the organic compounds, electron acceptors and metabolic by-products have been monitored for 2 years—between September 2001 and September 2003 (Klonowski et al. 2005). Based on the monitoring data, the major factors controlling the development of the plume have been recognised and evaluated. The available data, however, does not allow a detailed interpretation of the rates of various processes. This study aims at estimating the contribution of biodegradation to the overall process of plume development and its natural attenuation. This, in turn, enables a long-term prediction of the contaminant degradation and plume behaviour.
Materials and methods
Site description
The research has been conducted at the firefighting training site of the international Oslo airport—Gardermoen, located about 50 km northeast of Oslo. The studied site used to belong to a military airport. Construction of a new civil airport, completed in 1998, has intensified firefighting training activities. At present, the site is used to train the firefighters of the National Aviation Authorities from the whole country. Potential spills of the jet fuel, used for training purposes, should be retrieved by a run-off collection system. Nevertheless, in 1998 a hydrocarbon contamination originating from a leaking oil skimmer, a part of the collecting system, was found in the soil and groundwater (Rudolph-Lund and Sparrevik 1999).
The Gardermoen airport is located on an ice contact glacio–marine fan delta. This was deposited in a shallow marine environment during the early Holocene phase of the Scandinavian ice cap retreat (Soerensen 1979). The Gardermoen fan delta consists of three units, namely: topset, foreset and bottomset. At the studied area, the topset unit is formed by about 2 m thick, layer of gravels and coarse sands. This is underlain by the foreset unit consisting of dipping southwest, interbeded layers of fine and coarse sands. The bottomset unit consists of fine sands, silts and clays.
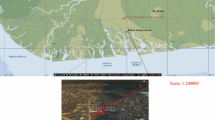
The Gardermoen delta forms a phreatic aquifer, recharge mainly—about 60%, by snow melting (Joergensen and Oestmo 1992). The aquifer is divided into two catchments areas and the groundwater divide, striking NNW–SSE, stretches out throughout the middle of the airport. Northeast of the groundwater divide the groundwater flows towards a number of the kettelhole lakes and the Risa river, while southwest of the divide, including the studied area, the groundwater is drained via the numerous springs into the Sogna and Vika rivers (Oestmo 1976). This situation is illustrated in Fig. 1. Significant groundwater table fluctuation, reaching up to a 2-m range, has been observed between September 2000 and September 2003 (Klonowski et al. 2005). Hydraulic conductivity of the deposits ranges between 6.63·10−5 and 2.11·10−2 (m · s−1), and reaches maximum at the depth of about 4 m and decreases thereafter with depth (Klonowski et al. 2005). Fig. 1 illustrates geological settings as well as some hydrogeological and hydrological features of the area adjacent to the studied site.

A network of nine piezometers, referred to as P1–P6 and P8–P10, and two observation wells, referred to as Br1 and Ano11, has been installed in order to monitor elevation of the groundwater table. A network of 16 multilevel samplers, referred to as: MLS 1–MLS 13, MLS 15–MLS 16 and MLS 19, has been installed in order to monitor groundwater quality and concentration of soil gases. The code for each multilevel sampler consists of the sampler number and a sampling port number, e.g. MLS 10/3. The numbering of the sampling ports starts at the bottom. The samplers cover both unsaturated and saturated zones, up to a depth of about 12 m. In addition, installation of some multilevel samplers enabled sediments sampling (Klonowski et al. 2005). The localisation of the selected monitoring points is illustrated in Fig. 2.

Localisation of the monitoring network: a map of the studied area and b longitudinal cross-section SW–NE (MLS 4–MLS 8)
Hydrogeochemical settings
The hydrocarbons plume, resulting from the leaking oil skimmer, has been detected in the topset and foreset units, in both saturated and unsaturated zones (Rudolph-Lund and Sparrevik 1999). The leaking skimmer has been dismantled and replaced by the observation well Br1 which is further on referred to as a contamination source zone. A part of the hydrocarbons free phase and contaminated groundwater was removed during the construction work. In the subsequent years the plume was a subject to the detailed geochemical and hydrogeological studies. Below, only the most important features of the studied plume are given—for the details please refer to our earlier work (Klonowski et al. 2005). The sampled sediments contain the jet fuel components: n-C10–n-C16. The highest concentrations, up to 3,900 (mg · kg−1) of dry weight sediment, was found along the whole sampled profile at the source zone, while downgradient hydrocarbons were constrained to a narrow zone of 4–5 m deep. The plume of the organics detected in the sediment samples is at least 17 m long. The hydrocarbons have been smeared out in vertical direction due to the groundwater table fluctuation.
The following dissolved hydrocarbons were found in the groundwater samples: toluene, ethylbenzene, o-xylene, m/p-xylene, 1,2,4-trimethylbenzene (1,2,4-TMB), 1,3,5-trimethylbenzene (1,3,5-TMB) and naphthalene. The zone of high concentration of the dissolved hydrocarbons, referred to as a core of the plume, coincided with the maximum concentrations of the hydrocarbons determined in the sediment samples. The concentrations of the individual jet fuel components differed within the plume. Toluene showed very small concentrations limited to the source zone and its close vicinity, while xylenes and ethylbenzene formed the larger plumes and show higher concentrations. Naphthalene was found the most recalcitrant—it showed the highest concentrations and formed the most extensive plume. The concentrations of the dissolved hydrocarbons showed noticeable variation over time caused by the groundwater table fluctuation.
The data on concentrations of the electron acceptors and metabolic by-products in the aquifer were used to study evidence of the natural attenuation processes. The depletion zones of the electron acceptors corresponded to the zone of high concentration of the dissolved hydrocarbons. Increased alkalinity of these zones indicated a biological character of the observed removal of the dissolved organics. The observed concentrations of the electron acceptors and metabolic by-products correlate well with changes in hydrocarbons concentration.
Experimental set up
The field experiment, a combination of a push–pull test and a tracer test, described in this paper was conducted under natural hydraulic gradient conditions between September and December 2003. The tracer solutes were prepared in two brand new and clean steel oil drums, further on referred to as drum one and drum two, with capacity of 200 L each, equipped with an electric stirrer and a tightly closed lid. The drums were completely filled with groundwater retrieved from the plume via the following sampling ports: MLS 10/2, MLS 12/2 and MLS 13/2, located just below the groundwater table. Before filling the empty drums up, those were flushed with a technical purity nitrogen—in order to avoid oxygenation of the groundwater. In addition, a slight nitrogen overpressure was maintained during mixing and injection of the tracer. The groundwater stored in the drums was spiked with a mixture of the organic compounds found in the studied plume: toluene, o-xylene, 1,2,4-TMB, 1,3,5-TMB and naphthalene. The mass ratio of these compounds was chosen as 1:2:2:1 (toluene:o-xylene:total trimethylbenzenes:naphthalene), based upon the concentration of the aromatic hydrocarbons dissolved in the deionised water in contact with pure jet fuel (Breedveld et al. 1997). Naphthalene, as a solid, was dissolved first in the remaining liquid organic compounds. After about 3-h stirring, the groundwater was spiked with a solution of potassium bromide. To ensure the proper mixing of the solutes the tracer was continuously stirred prior to and during the injection, for approximately 12 h. Identical procedures were applied to both drums.
Concentration of all the tracer solutes was maintained high enough to yield detectible concentrations in the whole plume, taking into account dilution, dispersion and sorption, even as far as the MLS 4—about 37 m downgradient from the injection points. The concentrations were 3,000 and 60 (mg · L−1) for total organic compounds and potassium bromide, respectively. The compounds, of analytical grade and purity, were purchased from VWR International (VWR International, West Chester, USA) and SIGMA-ALDRICH (Sigma-Aldrich, St. Luis, USA). Selected physical and chemical properties of the tracer components are given in Table 1 (Lyman et al. 1990; Montgomery 2000), while mean concentrations of the solutes in the tracer solution and their statistical moments are given in Table 2.
The tracer was injected into the aquifer via three separate injection points—MLS 10/3, MLS 12/3 and MLS 13/3, situated 0.5-m above the groundwater extraction points. A three channelled peristaltic pump Watson and Marlow, model 505S (Watson and Marlow Ltd., Falmouth, Great Britain), was first used to fill the drums up with the groundwater and then to inject the tracer. The pumping rate applied in both cases was kept at a constant level of about 0.25 L per minute, for all three channels together. In order to minimise sorption of the organic compounds the Teflon and tygon tubing was used. The tracer solution was injected over a 24-h period—starting on 9 October, 20.00 p.m. A total volume of 370 L was injected (entire drum one and 170 L of drum two). The tracer samples were collected during injection—twice from drum one and three times from drum two. Concentrations of the tracer solutes and major ions were relatively stable during injection, though some small differences between both drums were found. Oxygen concentration remained stable in the drum one, about 2 (mg · L−1), however it increased up to more than 7 (mg · L−1) in the drum two, as a result of nitrogen gas depletion. Bicarbonate and Fe(II) showed slightly decreasing trends. Nitrate was not detected in the tracer solution at all. Small concentrations of ethylbenzene and m/p-xylene detected in the tracer solution reflect the composition of the contaminated groundwater used for preparation of the tracer.
A network of multilevel samplers allows groundwater and soil gas sampling from the different locations and depths within the studied area. In order to minimise sorption processes the stainless steel mesh and Teflon tubing (inner diameter 1/8 in) were used for constructing the samplers. The sampling ports were spaced at 0.5 m of vertical intervals and covered both unsaturated and saturated zones.
The groundwater sampling frequency varied during the experiment. The groundwater was sampled twice per day when the most significant changes occurred—at the beginning of tracer removal from the injection points and at tracer arrival at the observation points. Subsequently, the groundwater was sampled once per day, twice per week and once per week. The total duration of the experiment was about 60 days (1,500 h).
Field measurements, sampling procedures and laboratory analyses
Determination of the following groundwater parameters: pH, electroconductivity, temperature and dissolved oxygen content, were conducted using the field instruments and a flow-through cell, prior to sampling. The following instruments were used—for pH determination Orion, model 250A (Orion Research Inc., Boston, USA), for determination of electroconductivity and temperature WTW Cond, model 330i (WTW, Weilheim, Germany) and for determination of oxygen content Oxi, model 330 (WTW, Weilheim, Germany). The groundwater samples were collected using a multichannel peristaltic pump—Ismatec (Ismatec SA, Zurich, Switzerland).
Groundwater sampling started after stable readings of the measured groundwater parameters were reached. This usually took about 5–10 min pumping at the average rate of 60 mL per minute, purging approximately 0.5 L of the groundwater. The pump was directly connected to a set of tightly capped glass bottles. Such procedure, called in-line sampling, minimises exposure of the groundwater to atmospheric oxygen. All the groundwater samples were stored in a dark place at ambient temperature and transported to the laboratory on the sampling day.
The samples for analyses of concentration of the dissolved hydrocarbons were collected into the 120 mL glass bottles, tightly capped with the Teflon lined silicon septa and stored upside down to minimise volatilisation. The concentrations of the dissolved hydrocarbons were quantified on the pentane extractions of the groundwater samples on the day of sampling. Usually 2 mL, in rare cases 3 mL of pentane, with 1-Cl-4F benzene as an internal standard, have been used. The extracts were analysed by a Chrompack CP9001 gas chromatograph (Chrompack, Mideelburg, the Netherlands) equipped with a split/splitless injector, CP-Sil 8-CB column (25 m×0.25 mL i.d.×1.2 μm) and a flame ionization detector. Based on the volumes of the sample and extractant, the detection limit was approximately 0.1 (μg · L−1) for each hydrocarbon and the accuracy is within 20%.
Alkalinity of the groundwater samples was determined in the field, immediately after sampling, by the direct Gran titration method (Appelo and Postma 1996; Stumm and Morgan 1996). The 30 mL groundwater samples were titrated by 0.01 normal hydrochloric acid up to pH of about 4.2, using a digital burette (Rudolf Brand GMBH+Co, Wertheim, Germany). The pH measurements were taken by the above-mentioned Orion instrument. Alkalinity was assumed to equal the concentration of HCO −3 , since the groundwater pH was below 8.3.
The groundwater sampled for determination of the major ions was filtered through a 0.45 μm filter tightly fitted into the in-line sampling system and collected in the 27 mL glass bottles. Concentration of the major anions: F−, Cl−, NO −3 and SO 2−4 was analysed by an ion chromatograph Dionex QIC (Dionex, Sunnyvale, USA). The groundwater samples taken for determination of major cations: Na+, K+, Ca2+, Mg2+, Fe2+ and Mn2+ were conserved in the field with 3-normal nitric acid, stored in a cooling room and analysed after the completion of the experiment using an atomic adsorption spectrograph Varian SpectrAA-300 (Varian Techtron, Springvale, USA).
Concentration of the soil gases were determined by a direct reading field instrument Multiwarn II (Dräger, Lübeck, Germany) measuring concentration of oxygen, carbon dioxide and methane in volume percentage. The instrument was connected to the sampling ports located in the unsaturated zone via a water trap. The measurements were taken three times, prior to the experiment, on the 20th of September 2003, during the experiment, on the 24th of October 2003, and after termination of the experiment, on the 12th of February 2004.
Data processing
Removal of the tracer solutes and breakthrough data were analysed by a method of moment analysis (Levenspiel 1989). The mean times, \( \overline{t} , \) of injection, removal and breakthrough are the first moments of the concentration versus time data. The moment analysis was conducted according to the linear interpretation method for the pulse response data separated by unequal time intervals, where C i and t i are the concentration and time at interval i, respectively:
Sorption implies partitioning of a chemical between the groundwater and the aquifer solids and results in retardation of a sorbed compound (Freeze and Cherry 1979; Karickhoff 1984). A retardation factor R can be used to express a ratio of groundwater transport and solute transport:
Where V w is an effective groundwater velocity, equal to the velocity of the non-reactive tracer, and V c is the mean velocity of the retarded solute. In case of the push–pull test, removal was observed and the retardation factors were calculated by dividing the mean removal time of the individual organic solute, \( \overline{t} _{{{\text{org}}}} , \) by the mean removal time of the non-reactive solute, bromide, \( \overline{t} _{{{\text{Br}} - }} , \) as:
Assuming linear, reversible and instantaneous sorption, the sorption distribution coefficient—K d, can be calculated from the observed retardation as follows:
Where ρb is bulk density of the aquifer solids (g · cm−3) and θ is the porosity of the aquifer (dimensionless fraction).
For comparison, sorption distribution coefficients have been also calculated based on the organic carbon content in the sediments samples (Karickhoff 1981):
and
Where K oc is a distribution coefficient of an organic compound between organic carbon and water, K ow is a distribution coefficient between water and octanol and f oc is a fraction of organic carbon in the sediment sample.
Determination of bulk density and porosity was conducted for the undisturbed sediment cores obtained from the Gardermoen aquifer at the Moreppen research field station by Knudsen (2003). Like the studied site, the Moereppen station is located in the distal part of the delta, about 1,000 metres north of the studied area (Fig. 1). Determination yielded the following results: ρb=1.68 (g · cm−3) and θ=0.367 (−) (Knudsen 2003), which were used for calculations in this paper.
The mean injection concentrations were determined by dividing the area under the concentration versus time curves by the injection time—24 h (Mackay et al. 1994). The removal and breakthrough data have been normalised by dividing the observed concentrations—C, by the mean injection concentrations—C0, in order to facilitate comparison of the individual monitored species.
The average linear groundwater flow velocity—V x , also called seepage velocity or pore water flow velocity, was calculated according to the following equation (Appelo and Postma 1996; Fetter 2001):
Where K is hydraulic conductivity, θ is effective porosity and dh/dl is hydraulic gradient. The hydraulic gradient was obtained from the groundwater table elevation data.
Effective groundwater flow velocity was calculated for the field experiment by dividing the distance between the injection point and the monitoring point by the mean travel time of the non-reactive solute. This, in turn, was calculated as a difference between the mean arrival time and the mean injection time of the non-reactive tracer.
The plots illustrating two dimensional (2-D) distribution of the selected electron acceptors and metabolic by-products were prepared in Surfer software, version 8.0, using kriging method for data interpolation (Golden Software 2002). An anisotropy ratio of 1:10 was used, based on the previous studies conducted at the Moereppen research station (Alfnes et al. 2003a, b; Knudsen 2003).
Results
Hydrogeological settings
Prior to and during the experiment, the groundwater table elevation was monitored in the piezometer P8 located nearby the plume. Summer and autumn 2003 were rather dry and relatively deep groundwater table position was expected. The measurements showed that the groundwater table was constantly decreasing during the experimental period—namely, between the 22nd September and 12th December 2003, the groundwater table elevation dropped from 198.89 to 198.70 m above sea level.
The hydraulic gradient of the studied aquifer was determined three times during the experimental period, for the piezometers P2 and P10. It yielded 0.0043 for each calculation. Assuming hydraulic conductivities calculated according to the Gustafson method (Klonowski et al. 2005) and porosity equal to 0.367 (Knudsen 2003) the average linear groundwater velocities were calculated for the injection and monitoring points. The results are presented in Table 5.
Initial hydrogeochemical conditions
In order to determine the hydrogeochemical conditions under which the experiment was conducted an initial groundwater sampling session was carried out prior to the experiment. Totally, 49 groundwater samples were collected for determination of the major inorganic species and 38 for determination of the organic compounds. The samples were obtained from all the sampling ports of the following multilevel samplers: MLS 10, 12, 13, 11, 9, 3, 15, 9 and 4. The observed distribution of the dissolved hydrocarbons as well as the inorganic species were comparable to those observed in the previous sampling sessions (Klonowski et al. 2005), therefore only the main features are given below.
The plume of the dissolved hydrocarbons was detected in about 1-m thick top zone of the aquifer. Naphthalene was the main constituent—it formed the most extensive plume and revealed the highest concentration, up to about 700 (μg · L−1) at MLS 10/2—1.1 m downgradient from the source zone. The concentration decreased gradually downgradient and at MLS 19/1—37.5 m downgradient of the source zone, reached 27 (μg · L−1), being the only hydrocarbon detected at this point. The plumes of 1,2,4-TMB and 1,3,5-TMB showed a similar extent, of about 17 m, however concentrations of 1,2,4 TMB were relatively higher. Toluene and o-xylene were detected only in small amounts, close to the source zone, while ethylbenzene and m/p-xylene showed some intermediate features. Distribution of the selected individual jet fuel components determined for the plume core along the longitudinal cross-section MLS 10–MLS 4 is illustrated in Fig. 3.

Spatial distribution of individual dissolved hydrocarbons for the longitudinal cross-section MLS 10–MLS 19 determined for the September 2003 sampling event
Spatial analysis of the electron acceptors and metabolic by-products revealed a distinct zonation, strongly correlated with distribution of the dissolved hydrocarbons. A zone of depleted dissolved oxygen was observed along the whole plume. Minimum concentrations—about 1 (mg · L−1) and below, coincided with maximum concentration of the dissolved hydrocarbons at MLS 10 and MLS 11—1.1 and 3.2 m downgradient from the source zone, respectively. In the plume’s outskirts slightly higher concentrations were observed. Nitrate was almost completely depleted at the plume core. Increased concentration of Mn(II)—up to about 3 (mg · L−1), were limited to the top zone of the aquifer at a distance of 7.5 m downgradient of the source zone. In contrast, increased Fe(II) concentrations—around 100 (mg · L−1) and higher, were found within most of the plume—up to about 17 m downgradient of the source zone. A zone of sulphate depletion was found to strictly coincide with the core of the hydrocarbons plume. A zone of increased bicarbonate concentrations was found within most of the plume—up to MLS 15, 17 m downgradient of the source zone. Its maximum values coincided with maximum hydrocarbons concentration. Bicarbonate and sulphate concentrations showed also an increase with depth due to pyrite and calcite dissolution. These processes were observed earlier in the Gardermoen aquifer (Basberg et al. 1998; Klonowski et al. 2005; Knudsen 2003). Spatial distribution of the electron acceptors and metabolic by-products, for the longitudinal cross-section MLS 10–MLS 4, showing the initial geochemical conditions of the experiment is illustrated in Fig. 4.

Spatial distribution of: a total dissolved hydrocarbons, b O2, c NO −3 , d Fe2+, e Mn2+, f SO 2−4 and g HCO −3 for the longitudinal cross-section SW–NE (MLS 4–MLS 10)
Push–pull test
The push–pull test was based on monitoring of the tracer solutes removal and induced changes in groundwater geochemistry at the injection points—MLS 10/3, MLS 12/3 and MLS 13/3, located 1.1, 1.5 and 1.5 m downgradient from the source zone, respectively. About 85 h after injection started, the groundwater table dropped below the injection point MLS 13/3. Due to this fact, the full tracer removal data sets were obtained only for MLS 10/3 and MLS 12/3. Normalised concentration of the selected organic compounds, electron acceptors and metabolic by-products, versus time for the injection point MLS 10/3, are illustrated in Fig. 5. Normalised concentration of bromide for some observations exceeded one which might be a result of analytical error for the high concentration range.

Changes in time of normalised concentrations of organic and inorganic solutes for the injection point MLS 10/3: a Br−, toluene, o-Xylene, 1,2,4-TMB, 1,3,5-TMB and naphthalene, b O2, SO 2−4 , and c HCO −3 , Fe2+, Mn2 +
Removal of the non-reactive bromide solute was very rapid and noticeably higher for MLS 12/3 than for MLS 10/3. This is due to the difference in hydraulic conductivity, which for the earlier, is 9.58·10−4 (m · s−1), while for the latter is about 2.71–3.75·10−4 (m · s−1) (Klonowski et al. 2005). Compared to bromide, removal of the reactive organic solutes from the injection points was retarded—toluene was removed first, than o-xylene and finally the remaining compounds. Between the 114th and 138th hour of the experiment a significant fluctuation of organic compounds concentrations was observed, which will be discussed below.
Injection and subsequent removal of the tracer solutes induced changes in groundwater chemistry. A very strong fluctuation of dissolved oxygen concentration was observed for the injection points 10/3 and 12/3. First, the fluctuation range was rather small, below the concentration in the tracer, while after about 180 h much higher variations were observed. Sulphate concentration, for MLS 10/3, revealed first an abrupt increase—from C·C −10 =1 up to almost 3.5 and then a gradual decrease since about the 180th hour onward. For MLS 12/3 a gradual decrease from about C·C −10 =1 to about 0.5 during the experiment was observed. Patterns of Mn(II) and Fe(II) concentrations resembled each other. At MLS 10/3 a constant decrease and a short period of stabilisation was observed first. The concentrations increased suddenly since about the 300th hour and stabilised towards the end of the experiment. In turn, for MLS 12/3 a very steep increase was detected until about the 200th hour of the experiment and then a slight decrease and stabilisation of concentrations. Bicarbonate concentration for MLS 10/3 increased until about the 140th hour and decreased for the remaining part of the experiment. For MLS 12/3 only a decrease was observed. Different concentration patterns, especially for Mn(II) and Fe(II), observed for both injection points might have been an effect of different removal rates. The changes observed at the beginning of the observations at MLS 10/3 might also occurred for MLS 12/3 before the observation started.
Because nitrate was not detected in the tracer at all, it is not shown in Fig. 5. Nitrate was detected in the groundwater only at MLS 10/3. Its absolute concentrations ranged between 4.25 and 0.31 (mg · L−1), with maximum at the beginning of the experiment and gradual decrease over time.
Tracer test
The aim of the tracer test was to examine the transport of the tracer solutes and induced changes of groundwater chemistry. Tracer transport was monitored at the sampling ports located 0.5 m below the injection points—MLS 10/2, MLS 12/2 and MLS 13/2 as well as downgradient—at the MLS 11, MLS 9 and MLS 15—3.2, 7.5 and 17.0 m of the source zone, respectively. The distance between the injection point MLS 10/3 and the monitoring point MLS 11/3 was 2.1 m.
At the monitoring point MLS 10/2 only a very small part of bromide solute was detected—maximally 0.0047 of the normalised concentration, at the 135th hour of the experiment. Maximum concentration of the reactive solutes occurred at the same time. Naphthalene revealed the highest concentration—up to 0.18 of normalised concentration, while 1,2,4-TMB, 1,3,5-TMB, o-xylene and toluene showed 0.1, 0.04, 0.01 and 0.006, respectively. Further on, at the 516th hour, a single peak for all organics was detected. Concentrations of the electron acceptors and metabolic by-products remained rather stable during most of the experiment. Some slightly increased values of bicarbonate and decreased levels of oxygen were observed at the beginning of the experiment. None of the tracer solutes was detected at the monitoring point MLS 10/1. No nitrate was detected in monitoring point MLS 10/2 and small concentrations—up to 1.15 (mg · L−1), were determined for MLS 10/1.
Very similar concentration patterns of the tracer solutes, electron acceptors and metabolic by-products were found for the monitoring points MLS 12/2 and MLS 13/2. For both very low bromide concentrations, 0.0001 of normalised concentration, were detected at the beginning of observations—at the 88th hour. Those were followed by the organic solutes, however concentrations for MLS 13/3 were slightly higher then for MLS 12/2. For example, relative concentration of naphthalene reached up to 0.0526 for the earlier and up to 0.014 for the latter, maximally. Concentrations of o-xylene and toluene were up to one order of magnitude lower. Concentrations of the electron acceptors and metabolic by-products remained rather constant during the observation period. Some variations in oxygen concentration and depletion of sulphate at the beginning of the experiment were noticed. No nitrate was detected in monitoring point MLS 12/2 and MLS 13/2.
The breakthrough curves for the monitoring point MLS 11/3 and concentrations of the selected electron acceptors and metabolic by-products are illustrated in Fig. 6. Bromide was detected after 85 h of observations and lasted up to the 235.5th hour. Maximum normalised concentration reached slightly more than 0.21 at the 132th hour of the experiment. The peak concentrations of the organic compounds were observed between the 110th and the 161st hour, reaching up to: 0.1211, 0.0546 and 0.0153 of normalised concentration for naphthalene, 1,2,4-TMB and 1,3,5-TMB, respectively. No increase in neither o-xylene nor toluene concentrations above the initial conditions was detected. No retardation of the organics was observed. After the peak, the tracer solutes concentrations decreased to the level of initial conditions, detected prior to the experiment and remained stable. The results of the moment analysis for the breaking through solutes at the MLS 11/3 and the calculated residence times for migration from MLS 10/3 are given in Table 3.

Breakthrough curves for the monitoring point MLS 11/3: a Br−, 1,2,4-TMB, 1,3,5-TMB and naphthalene, b O2, HCO −3 , Fe2+, Mn2 +
An appropriate response in the electron acceptors and metabolic by-products concentrations was observed. A peak of Fe(II) and Mn(II) concentrations, coinciding with the peak of the tracer solutes, was found. The maximum normalised concentrations amounted up to three and four for Fe(II) and Mn(II), respectively. After the peak, the concentrations showed a slight increasing trend with time. Oxygen concentrations were rather unstable and fluctuated significantly, especially between the 110th and 156th hour. A significant increase was observed for two last measurements. Bicarbonate concentrations showed first a decrease at the arrival of the tracer and then a sudden increase. Afterwards, the concentration showed a slight increasing trend over time. Neither nitrate nor sulphate was detected.
At the monitoring point MLS 11/2 very low concentrations of the tracer solutes were detected. Bromide was observed at the beginning of the observations, between the 114th and the 183rd hour, with the maximum normalised concentration of 0.0075. Out of all the injected organic solutes only naphthalene and 1,2,4-TMB appeared, with the maximum normalised concentrations up to 0.03 and 0.02, respectively. Those compounds showed a stable increase of concentration during the experiment. Oxygen concentration was rather unstable. Concentration of bicarbonate revealed slightly higher values coinciding with the tracer peak, and then a decrease. The same patterns were observed for sulphate, Mn(II) and Fe(II). Nitrate was not detected at all.
Groundwater sampling at the multilevel samplers located further downgradient—MLS 9 and MLS 15 did not yield any breakthrough results. No bromide, organic solutes nor changes in electron acceptor and metabolic by-products concentration were detected.
Soil gases concentration
The measurements of carbon dioxide and oxygen concentrations in soil atmosphere were carried out to assess carbon dioxide production resulting from biodegradation of hydrocarbons and to indirectly evaluate the natural attenuation processes. First soil gas monitoring session, conducted on the 20th of September 2003, determined the initial conditions related to the developed hydrocarbons plume. The next two sampling sessions took place during and after the experiment, on the 24th of October 2003 and the 12th of February 2004, respectively. Those were carried out in order to evaluate short- and long-term effect of the injected tracer on carbon dioxide production. The results of these three monitoring sessions, determined for the sampling ports located directly above the groundwater table, are illustrated in Fig. 7. Measurements taken at MLS 8 represent the background values. Constant increase in carbon dioxide concentrations in time for all the monitored sampling ports was found. The highest detected concentrations coincided with the hydrocarbons source zone. An abrupt decrease downgradient of the source zone was observed. Oxygen concentrations showed the expected opposite patterns. Two first sampling sessions were conducted in autumn—under very similar weather and vegetation conditions and an open system with respect to oxygen and carbon dioxide diffusion. The third session was carried out in winter, when carbon dioxide production in the root zone was minimised and the snow cover and frozen ground formed a closed environment preventing replenishment of oxygen and diffusion of carbon dioxide (Appelo and Postma 1996). These effects might have influenced the obtained results.

Concentration of a CO2 and b O2 in the soil atmosphere for the sampling ports located directly above the groundwater level along the cross-section MLS 8–MLS 19
Solute transport parameters
Retardation in removal of the individual organic compounds was detected for the push–pull test. Correlation between the retardation rate of the injected organics and their properties, like hydrophobicity and affinity to organic carbon, was observed—first toluene was removed, than o-xylene and subsequently the remaining compounds. Retardation factors and sorption distribution coefficients were calculated based on the moment analysis and theoretically based on the content of organic carbon in the sediments samples. The results are given in Table 4 and Fig. 8.

Cross-plot showing sorption distribution coefficients for MLS 10/3 (small symbols) and MLS 12/3 (large symbols), single asterisk estimated for organic carbon fraction according to (Karickhoff 1981) and double asterisk calculated for the push–pull test
No retardation of the organics was detected for the monitoring point MLS 11/3. Based on the moment analysis of the bromide breakthrough data, the groundwater flow velocities were calculated for the monitoring points MLS 11/3 and 11/2. For comparison, the average groundwater velocities based on porosity, hydraulic conductivity and hydraulic gradient were calculated as well. The results are given in Table 5.
Biodegradation potential
In order to evaluate contribution of biodegradation to the overall process of natural attenuation of injected organic solutes, a budget of the electron acceptors and donors was accomplished. The following processes were taken into account: oxygen consumption, nitrate reduction, Mn(IV) reduction, Fe(III) reduction and sulphate reduction as well as degradation of injected organics: toluene, o-xylene, 1,3,5-TMB, 1,2,4-TMB and naphthalene. No data on methane production were collected.
Calculations were completed for the monitored transport between the injection point MLS 10/3 and the monitoring point MLS 11/3. The injected tracer solutes were a subject to the complex processes, including degradation, sorption, dilution and mixing with in situ groundwater. Concentrations determined for the injection point MLS 10/3 at the mean removal time of the total dissolved hydrocarbons, the 170th hour of the experiment, were assumed to represent these processes and were used as the starting point for biodegradation potential calculations. For the monitoring point MLS 11/3 the concentrations determined for the peak values were used. Calculations revealed that Fe(III) and sulphate reductions are the major factors controlling degradation of the organic solutes. Comparison of total available electrons, 6.73 (mmol · L−1) and total electrons demand −8.62 (mmol · L−1), showed that the observed electron accepting processes are capable to degrade the majority of the injected organics. Thus complete loss of toluene and o-xylene can be accounted for intrinsic biodegradation. The calculation results as well as the half-cell reactions for the electron acceptors and electron donors are given in Table 6.
Discussion
Hydrogeological and geochemical settings
The experiment was conducted within a strongly heterogeneous aquifer. Significant influence of dipping sedimentary structure of the Gardermoen delta foreset on the groundwater flow patterns, in both saturated and unsaturated zones, was reported in the previous research (Alfnes et al. 2003a, b; French 1999; Knudsen 2003; Sabir 2001; Soevik et al. 2002). Therefore, low concentrations of bromide, detected at the monitoring points, can to a great extent, be explained by preferential groundwater flow. The collected data set does not allow estimation of contribution of such process into the overall attenuation process. On the contrary, 0.5 m of vertical interval between the sampling ports might have been too large to monitor the migration of solutes in such a heterogeneous aquifer.
The tracer solutes were injected into the well-established system of biologically mediated redox reactions of hydrocarbons degradation (Klonowski et al. 2005). The geochemical conditions were changed upon tracer injection due to, among others, mixing of tracer solutes with in situ groundwater, dilution of tracer solutes, sorption and biodegradation.
Soil gas measurements
Spatial analysis of carbon dioxide and oxygen concentrations in the soil atmosphere showed that a surplus of carbon dioxide produced in the redox reactions of hydrocarbon degradation was released into the soil environment. After tracer injection, a substantial increase of carbon dioxide concentration in soil gas was detected. This fact supports hydrogeochemical data indicating that biological processes are the major cause of hydrocarbons removal.
Push–pull test
The data for the push–pull experiment were obtained by sampling groundwater via the injection points. Monitoring of the tracer solutes concentrations in time revealed quick removal of bromide and retardation of the organics which is illustrated by the results of mean removal time calculations. Removal was greater for the MLS 12/3 than for the MLS 10/3 because of higher permeability of the sediments and higher groundwater flow velocity for the earlier. In addition, due to high inflow of the groundwater from the background more electrons were available resulting in higher biodegradation potential and quicker removal of the organics. Whereas initial contamination detected prior to the experiment for MLS 12/3 was much higher than for MLS 10/3—for example for naphthalene by two orders of magnitude. This fact must affect the processes of sorption and degradation of the injected organic solutes.
The observed differences in removal of the individual organic compounds resulted from differences in their chemical and physical properties. Toluene and o-xylene, showing K oc and K ow values lower than trimethylbenzenes and naphthalene, were readily removed at both monitored injection points. Removal of trimethylbenzenes and naphthalene was expected to be more retarded. The correlation was confirmed by calculations, based on the push–pull test data, of sorption distribution coefficients—K d and retardation factors—R. Calculations based on the content of organic carbon in the sediments (Karickhoff 1981) showed similar correlation for K d and R values. Similar values of sorption distribution coefficients and retardation factors, of the same order of magnitude, were found for the sediment samples from the same site (Zheng et al. 2002) and for other locations at the Gardermoen aquifer (Alfnes et al. 2003a, b; Soevik et al. 2002).
The electron acceptors and metabolic by-products concentrations showed a response to injection of the organic solutes. Concentration patterns are similar for both monitored injection points and the differences can be explained by faster removal of the tracer solutes from MLS 12/3. Observed patterns of concentrations of the inorganic and organic species in time were, to some extent, a result of mixing the injected solutes with the groundwater. Nevertheless, the decreased concentrations of sulphate and increased bicarbonate can be interpreted as an effect of biodegradation. To be able to evaluate contribution of this mixing effect a detailed geochemical modelling is required.
Tracer test
Transport of the tracer solutes was monitored beneath the injection points and downgradient. The influence of the tracer was detected for the monitoring points MLS 10/2, 12/2 and 13/2, located 0.5 m below the injection points. Naphthalene was the major organic compound detected. The tracer solutes were not observed at the monitoring points MLS 10/1, MLS 12/1 and MLS 13/1 located 1 m below the injection points. The breakthrough of the tracer solutes was detected at the MLS 11/3—2.1 m downgradient from the injection point MLS 10/3, however the detected concentrations were low. No retardation of the organics compared to bromide was observed. This fact can be explained, to some extent, by the preferential groundwater flow. No peaks of toluene and o-xylene were detected which was attributed to biodegradation. Naphthalene was the most recalcitrant and yielded the highest concentrations. Recalcitrance of naphthalene under anaerobic conditions was observed for the sediment samples originating from the studied area in laboratory batch experiment (Zheng et al. 2001) and for other locations under similar conditions (Bjerg et al. 1999). Concentrations of Fe(II) and Mn(II) showed a clear increase coinciding with arrival of the tracer pulse indicating the biological character of the organics removal. Bicarbonate concentration at the MLS 11/3, upon arrival of the tracer, was observed to decrease and then increase rapidly. This might be an effect of waters mixing, though it could have had some microbiological reasons as well.
Biodegradation potential
The budget of the available electron acceptors and electron donors for degradation of the injected organic solutes between the MLS 10/3 and MLS 11/3 revealed that Fe(III) and sulphate reductions were the major processes contributing to biodegradation of the dissolved organics. No data on methane concentration were collected; however the earlier observations of the studied plume showed that methane production can be one of the major processes controlling biodegradation of the jet fuel hydrocarbons (Klonowski et al. 2005). Calculations showed that the total number of the available electron acceptors is high enough to achieve degradation of majority of the hydrocarbons. This was confirmed by the observed complete degradation of toluene and o-xylene and substantial decrease in concentrations of the remaining organic compounds.
Summary and conclusions
This paper deals with the field experiment combining the push–pull and tracer tests. The mixture of the conservative tracer—Br−, and the reactive organic solutes: toluene, o-xylene, 1,3,5-TMB, 1,2,4-TMB and naphthalene, were injected into the existing plume of the jet fuel derived hydrocarbons. The plume was developed in a strongly heterogeneous aquifer of the Gardermoen fan delta.
The results of the push–pull test showed very quick removal of the tracer solutes from the monitored injection points. Due to higher permeability of the aquifer matrix removal was faster for the injection point MLS 12/3 than for the point MLS 10/3. For both injection points a strong retardation in removal of the dissolved aromatics, in respect to non-reactive bromide, was observed as well as the differences in retardation between the individual organic compounds. Calculated sorption distribution coefficients K d and retardation factors R showed correlation between the removal rates and the physiochemical properties of the organics.
The tracer solutes were observed 0.5 m beneath the injection points and up to 2.1 m downgradient. At that distance from the injection points only Br−, naphthalene and trimethylbenzenes resulting from the injected pulse of the tracer solutes were detected. No retardation of the organics, compared to the conservative tracer, was observed which might be an effect of migration along some preferential flow path in the heterogeneous aquifer.
Concentrations of the electron acceptors and metabolic by-products responded to the injected organic solutes. Strong fluctuation of O2 concentrations are difficult to interpret, however changes in concentration of Mn2+, Fe2+, SO 2−4 and HCO −3 can be explained by biodegradation processes. Calculations of biodegradation potential revealed that the available amount of the electron acceptors is high enough to explain observed removal of toluene and o-xylene.
Nevertheless, it should be stressed that the calculated degradation potential is only an approximation of the processes. Other effects, like sorption, dilution, mixing of waters, volatilisation, etc., have to be taken into account. Geochemical modelling should be one of the most accurate methods to determine the role of the above controlling factors in the observed overall natural attenuation process.
References
Alfnes E, Breedveld GD, Kinzelbach W, Aagaard P (2003a) Investigation of hydrogeologic processes in a dipping layer structure. 2. Transport and biodegradation of organics. J Contam Hydrol DOI 10.1016/j.jconhyd.2003.08.006
Alfnes E, Kinzelbach W, Aagaard P (2003b) Investigation of hydrogeologic processes in a dipping layer structure: 1. The flow barrier effect. J Contam Hydrol DOI 10.1016/j.jconhyd.2003.08.003
Appelo CAJ, Postma D (1996) Geochemistry, groundwater and pollution, 3rd edn. A.A.Balkema, Rotterdam Brookfield
Barker JF, Patrick GC, Major D (1987) Natural attenuation of aromatic hydrocarbons in a shallow sand aquifer. Ground Water Monit R Winter: 64–71
Basberg L, Dagestad A, Engesgaard P (1998) Geochemical modelling of natural geo-/hydrochemical stratification dominated by pyrite oxidation and calcite dissolution in a glaciofluvial Quaternary deposit, Gardermoen, Norway. NGU bullet 434:10
Bennett PC, Siegel DE, Baedecker MJ, Hult MF (1993) Crude oil in a shallow sand and gravel aquifer—I.Hydrogeology and inorganic geochemistry. Appl Geochem 8:529–549
Bjerg PL, Rugge K, Cortsen J, Nielsen PH, Christensen TH (1999) Degradation of aromatic and chlorinated aliphatic hydrocarbons in the anaerobic part of the Grindsted Landfill leachate plume: in situ microcosm and laboratory batch experiments. Ground Water 37:113–121
Breedveld GD, Olstad G, Aagaard P (1997) Treatment of jet fuel-contaminated runoff water by subsurface infiltration. Bioremediation J 1:77–88
Chapelle FH (1999) Bioremediation of petroleum hydrocarbon-contaminated ground water: The perspectives of history and hydrology. Ground Water 37:122–132
Chapelle FH (2001) Ground-water microbiology and geochemistry, 2nd edn. Wiley, New York
Dagestad A (1998) In situairsparging as a remedial action at the Gardermoen aquifer, Southeast Norway (in Norwegian). PhD thesis, Norwegian University of Science and Technology, Trondheim, Norway
Eganhouse RP, Baedecker MJ, Cozzarelli IM, Aiken GR, Thorn KA, Dorsey TF (1993) Crude oil in a shallow sand and gravel aquifer-II. Organic geochemistry. Appl Geochem 8:551–567
Fetter CW (1992) Contaminant hydrogeology, 1st edn. Prentice Hall, London
Fetter CW (2001) Applied hydrogeology, 4th edn. Prentice Hall, London
Fitts CR (2002) Groundwater science, 1st edn. Academic, Amsterdam
Freeze RA, Cherry JA (1979) Groundwater, 1st edn. Prentice Hall, Englewood Cliffs
French HK (1999) Transport and degradation of deicing chemicals in a heterogenous unsaturated soil. PhD Thesis, Agricultural University of Norway
Golden Software I (2002) Surfer 8. User’s guide. Contouring and 3D surface mapping for scientists and engineers. Golden Software
Harvey AH (1998) Environmental chemistry of PAHs. In: Hutzinger O (ed) The handbook of environmental chemistry. Springer, Berlin Heidelberg New York, pp 1–54
Joergensen P, Oestmo SR (1992) Research programme. The environment of the subsurface. Part I: the Gardermoen project, 1992--1994. Introductory report. Literature review and project catalogue. Hydrogeology at Romerike
Karickhoff S (1984) Organic pollutant sorption in aquatic systems. J Hydraul Eng 110:707–735
Karickhoff SW (1981) Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils. Chemosphere 10:833–846
Klonowski MR, Breedveld GD, Aagaard P (2005) Natural attenuation of a jet fuel contaminated aquifer. Monitoring, field experimentation and geochemical mdeling. Spatial and temporal changes of jet fuel contamination in an unconfined sandy aquifer. PhD Thesis, University of Oslo
Knudsen JBS (2003) Reactive transport of dissolved aromatic compounds under oxygen limiting conditions in sandy aquifer sediments. PhD Thesis, University of Oslo
Levenspiel O (1989) Chemical reactor omnibook, 1st edn. OSU Book Stores, Inc., Corvallis
Lyman WJ, Reehl WF, Rosenblatt DH (1990) Handbook of chemical property estimation methods. Environmental behavior of organic compounds, 1st edn. American Chemical Society, Washington
Mackay DM, Bianchi-Mosquera G, Kopania AA, Kianjah H, Thorbjarnarson KW (1994) A forced-gradient experiment on solute transport in the Borden aquifer. 1. Experimental methods and moment analyses of results. Water Resource Res 30:369–383
Mackay DM, Freyberg DL, Roberts PV (1986) A natural gradient experiment on solute transport in a sand aquifer. 1. Approach and overview of plume movement. Water Resource Res 22:2017–2079
McAllister PM, Chiang CY (1994) A practical approach to evaluating natural attenuation of contaminants in groundwater. Ground Water Monit R Spring 161–173
Montgomery JH (2000) Groundwater chemicals desk reference, 3rd edn. Lewis Publishers, Boca Raton
Oestmo SR (1976) Hydrogeological map of Oevre Romerike; groundwater in loose sediments between Jessheim and Hurdalsjone—scale 1:20 000. Norwegian Geological Survey
Rudolph-Lund K, Sparrevik M (1999) Mapping of contamination spreading at the firefighting training place of the Gardermoen international airport (in Norwegian). Norwegian Geotechnical Institute
Sabir HI (2001) Transport of a de-icing chemical and synthetic DNA tracers in groundwater at Oslo airport, Gardermoen. Field experiments and numerical modelling. PhD Thesis, Agricultural University of Norway
Schroth MH, Kleikemper J, Bolliger C, Bernasconi SM, Zeyer J (2001) In situ assessment of microbial sulfate reduction in a petroleum-contaminated aquifer using push-pull tests and stable sulfur isotop analyses. J Contam Hydrol 51:179–195
Schwartz FW, Zhang H (2003) Fundamentals of ground water, 1st edn. Wiley, New York
Soerensen R (1979) Late Weichselian deglaciation in the Oslofjord area, south Norway. Boreas 8:241–246
Soevik AK, Alfnes E, Bredveld GD, French H, Pedersen TS, Aagaard P (2002) Transport and degradation of toluene and o-xylene in an unsaturated soil with dipping sedimentary layers. J Environ Qual 31:1809–1823
Stumm W, Morgan JJ (1996) Aquatic chemistry. An introduction emphasizing chemical equilibria in natural waters, 3rd edn. Wiley, New York
Thierrin J, Davis GB, Barber C (1995) A ground-water tracer test with deuterated compounds for monitoring in situ biodegradation and retardation of aromatic hydrocarbons. Ground Water 33:469–475
Wiedemeier TH, Rifai HS, Newell CJ, Wilson JT (1999) Natural attenuation of fuels and chlorinated solvents in the subsurface. Wiley, New York
Zheng Z, Aagaard P, Bredveld GD (2002) Sorption and anaerobic biodegradation of soluble aromatic compounds during groundwater transport. 1. Laboratory column experiments. Environ Geol 41:922–932
Zheng Z, Bredveld GD, Aagaard P (2001) Biodegradation of soluble aromatic compounds of jet fuel under anaerobic conditions: laboratory batch experiments. Appl Microbiol Biotechnol 57:572–578
Acknowledgements
The first author is deeply grateful to the management and colleagues of the Polish Geological Institute for allowing a study leave in order to complete this research. Special thanks are due to the Donald Kuennen Foundation (the Netherlands) which provided the grants for the scientific books and software. The authors would like to thank Øyvind Kvalvåg, Mufak Naoroz and Alf Nielsen for their help with conducting the fieldwork and laboratory analyses. Assistance of the employees of the Gardermoen Airport—Jarl Øvstedal, Stig Moen and Knut Ringheim is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kłonowski, M.R., Breedveld, G.D. & Aagaard, P. Natural gradient experiment on transport of jet fuel derived hydrocarbons in an unconfined sandy aquifer. Environ Geol 48, 1040–1057 (2005). https://doi.org/10.1007/s00254-005-0042-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00254-005-0042-y